Packages integrated in Scipion
This is a dynamic list of methods grouped by package that are present in Scipion. Some of them might come from a development environment and soon will be released.
Short-cuts
Appion AreTomo Atomstructutils Atsas BSOFT Bamfordlab CCP4 Chimera ChimeraX Cistem Continuousflex CryoDRGN Cryoassess Cryoef Cryomethods Cryosparc2 Deepfinder Dynamo ESRF Eman2 Emfacilities Empiar Emxlib Fsc3d Gautomatch Grigoriefflab Imagic Imod Legacy Localrec Locscale Motioncorr NovaCtf Phenix Pkpd Powerfit_scipion Pyseg Relion RelionTomo Resmap Sidesplitter Simple Sphire Spider Susan Tomo Tomoj TomosegmemTV Tomotwin Topaz Warp Xmipp2 Xmipp3 Xmipptomo cryoCARE emantomo gCTF prody2 pwed pwem pyworkflowtests tomo3D tomovizPackages
 Appion
Appion
Available methods:
-
dogpicker (Particle picking)
- Protocol to pick particles in a set of micrographs using appion dogpicker.
Contributors:












AreTomo
Available methods:
-
tilt-series align and reconstruct
- Protocol for fiducial-free alignment and reconstruction for tomography.
Contributors:
 Atomstructutils
Atomstructutils
Available methods:
-
RMSD validate map
- Protocol to calculate the RMSD between all pairs of atom structures in a set of them. It calculates the overall RMSD for all of them and for each of their residues to validate their associated volume
-
average_sub_unit
- Average densities related with the given atomic structures. For example hexons in a unit cell. Alpha carbon are used to compute the transformation matrix
-
convert_sym
- In development
-
operator
- Utilities for handling PDB/mmcif atomic structure files.Current plugin utilities: (A) extract a chain from an atom structure (pdb/cif file),(B) perform union of several atomic structures
Contributors:



 Atsas
Atsas
Available methods:
-
convert PDB to SAXS curve
- Protocol for converting a PDB file (true atoms or pseudoatoms) into a SAXS curve. This is actually a wrapper to the program Crysol from Atsas. See documentation at: http://www.embl-hamburg.de/biosaxs/manuals/crysol.html
Contributors:

 BSOFT
BSOFT
Available methods:
-
bfilter
- Wrapper around bfilter program of BSOFT.
-
blocres (Local resolution)
- Bsoft program: blocres It calculates the local resolution map from to half maps. The method is based on a local measurement inside a mobile window.
-
particle picking (Particle picking)
- Protocol to pick particles in a set of micrographs using bsoft
Contributors:
 Bamfordlab
Bamfordlab
Available methods:
-
ethan picker (Particle picking)
- ETHAN is a program for automatic detection of spherical particles from electron micrographs. The ETHAN software was written at the Department of Computer Science of University of Helsinki, Finland by Teemu Kivioja.
Contributors:
 CCP4
CCP4
Available methods:
-
coot refinement
- Coot is an interactive graphical application formacromolecular model building, model completionand validation. IMPORTANT: press "w" in coot to transferthe pdb file from coot to scipion '
-
refmac
- Automatic refinement program in Fourier space of macromolecule structures regarding electron density maps. Generates files for volumes and FSCs to submit structures to EMDB
Contributors:
Chimera
Available methods:
-
alphafold prediction (Import)
- Protocol to import atomic structures generated by alphafold. If you choose the "Execute alphafold Locally" option you will need a local alphafold NO docker instalation as described here: https://github.com/kalininalab/alphafold_non_docker
Contributors:
 ChimeraX
ChimeraX
This plugin allows to use chimeraX commands within the Scipion framework.
Plugin url https://github.com/scipion-em/scipion-em-chimera.
Available in Linux. Scientific software, Image processing, cryo emAvailable methods:
-
alphafold prediction (Import)
- Protocol to import atomic structures generated by alphafold. If you choose the "Execute alphafold Locally" option you will need a local alphafold NO docker instalation as described here: https://github.com/kalininalab/alphafold_non_docker
-
contacts
- Identifies interatomic clashes and contacts based on van der Waals radii
-
map subtraction
- Protocol to subtract two volumes. One of these volumes can be derived from an atomic structure. Execute command *scipionwrite #n [prefix stringAddedToFilename]* from command line in order to transfer the generated maps and models to scipion. In addition to maps and models that the protocol saves by default, the user can generate and save some others
-
model from template
- Protocol to model three-dimensional structures of proteins using Modeller. Execute command *scipionwrite #n [prefix stringAddedToFilename] from command line in order to transfer the selected pdb to scipion. Default value is model=#0, model refers to the pdb file.
-
operate
- This protocol provides access to Chimera and allows to save the result in Scipion framework. Execute command *scipionwrite #n [prefix stringAddedToFilename] model refers to the pdb file
-
restore session
- This protocol opens Chimera and restores a session that has been stored each time a 3Dmap or an atomic structure by using `scipionwrite` or `scipionss` commad. Execute command *scipionwrite #n [prefix stringAddedToFilename] model refers to the pdb file
-
rigid fit
- Protocol to perform rigid fit using Chimera. Execute command *scipionwrite #n [prefix stringAddedToFilename] model refers to the pdb file
Contributors:
 Cistem
Cistem
Available methods:
-
classify 2D (2D classification)
- Protocol to run 2D classification in cisTEM.
-
ctffind4 (CTF estimation)
- Estimate CTF for a set of micrographs with ctffind4. To find more information about ctffind4 visit: https://grigoriefflab.umassmed.edu/ctffind4
-
find particles (Particle picking)
- Protocol to pick particles (ab-initio or reference-based) using cisTEM.
-
import tomo CTFs (Import)
- Protocol to import CTF estimation of a tilt-series from CTFFIND4.
-
tilt-series ctffind4 (CTF estimation)
- CTF estimation on a set of tilt series using CTFFIND4.
-
tiltseries ctffind4 (CTF estimation)
- CTF estimation on a set of tilt series using CTFFIND4.
-
unblur (Movie alignment)
- This protocol wraps unblur movie alignment program.
Contributors:


 Continuousflex
Continuousflex
Available methods:
-
MD-NMMD-Genesis
- Protocol to perform MD/NMMD simulation based on GENESIS.
-
NMMD refine
- Protocol to perform NMMD refinement using GENESIS
-
apply subtomogram alignment
- Protocol for subtomogram alignment after STA
-
classify subtomograms
- Protocol applying post alignment classification on subtomograms.
-
cluster set
- Protocol executed when a set of cluster is created from set of pdbs.
-
convert a PDB
- Convert a PDB file into a volume.
-
convert to pseudoatoms
- Converts an EM volume into pseudoatoms
-
deep hemnma infer
- This protocol is DeepHEMNMA
-
deep hemnma train
- DeepHEMNMA protocol, a neural network that learns the rigid-body parameters and the normal mode amplitudes estimated by HEMNMA protocol.
-
generate topology
- Protocol to generate topology files for GENESIS simulations
-
histogram matching
- Protocol for volume histogram matching.
-
missing wedge filling
- Protocol for subtomogram missingwedge filling.
-
missing wedge restoration
- Protocol for subtomogram missingwedge restoration.
-
nma alignment
- Protocol for flexible angular alignment.
-
nma alignment vol
- Protocol for rigid-body and elastic alignment for volumes using NMA. This protocol is the code module of HEMNMA-3D. It will take as input a set of normal modes calculated for an input atomic or pseudoatomic structure, and a set of volumes (subtomograms) to analyze. It fits the input structure using its modes (a subset of the modes need to be selected) into each one of the input volumes while simultaneously looking for rigid-body alignment, with compensation for missing wedge artefacts. The result of this protocol are rigid-body and elastic parameters for each input volume. Those results will be fed for a dimensionality reduction method (nma dimred vol) for further processing.
-
nma analysis
- Flexible angular alignment using normal modes
-
nma cluster
- Protocol executed when a cluster is created from NMA images and theirs deformations.
-
nma dimred
- This protocol will take the volumes with NMA deformations as points in a N-dimensional space (where N is the number of computed normal modes) and will project them onto a reduced space
-
nma vol cluster
- Protocol executed when a cluster is created from NMA volumes and theirs deformations.
-
nma vol dimred
- This protocol will take the volumes with NMA deformations as points in a N-dimensional space (where N is the number of computed normal modes) and will project them onto a reduced space
-
pdb dimentionality reduction
- Protocol for applying dimentionality reduction on PDB files.
-
pdbs rigid body alignement
- Protocol to perform rigid body alignement on a set of PDB files.
-
refine subtomogram alignment
- Protocol for refining subtomogram alignment and filling the missing wedge based on optical flow and Fast Rotational Matching (FRM). The protocol takes as input a set of subtomograms, with their subtomogram averaging protocol. It uses this global subtomogrm average to fill the missing wedge in Fourier space (the missing wedge is replaced by the corresponding region from the global average). Optical flow is used to match the global average with each of the missing wedge filled and aligned subtomograms (matched subtomograms are generated). Rigid-body alignment is performed using FRM from the matched subtomogram, and the rigid-body alignment for the input subtomograms is updated. Few iterations are usually sufficient (1-5), and the rigid-body alignment will be refined
-
structure mapping
- A quantitive analysis of dissimilarities (distances) among the EM maps that placing the entire set of density maps in to a common space of comparison.The approach is based on statistical analysis of distance among elastically aligned EM maps, and results in visualizing those maps as points in a lower dimensional distance space.
-
subtomogram averaging
- Protocol for subtomogram averaging. This protocol has two modes of operation. the first is to perform subtomogram averaging using Fast Rotational Matching. The second mode is to import a previously performed alignment using this protocol, Dynamo, or Artiatomi. If an alignment is imported, the rigid-body parameters will be used to re-create the average structure.
-
synthesize images
- Protocol for synthesizing images.
-
synthesize subtomograms
- Protocol for synthesizing subtomograms.
-
tomoflow dimred
- This protocol will take volumes with optical flows, it will operate on the correlation mat and will project it onto a reduced space
-
tomoflow protocol
- Protocol for HeteroFlow.
-
tomoflow vol cluster
- Protocol executed when a cluster is created from HeteroFlow dimred.
-
volume denoise
- Protocol for subtomogram missingwedge filling.
Contributors:





 CryoDRGN
CryoDRGN
Available methods:
-
preprocess (Continuous flexibility)
- Description missing, might be a new development.
-
training VAE (Continuous flexibility)
- Description missing, might be a new development.
-
training ab initio (Continuous flexibility)
- Description missing, might be a new development.
Contributors:
Cryoassess
This plugin provide a wrapper around Cryoassess software tools for automatic micrograph and 2D classes assessment.
Plugin url https://github.com/scipion-em/scipion-em-cryoassess/.
Available in Linux. Scientific software, Image processing, cryo emAvailable methods:
-
assess 2D classes
- Protocol to assess 2D classes and 2D averages
-
assess micrographs
- Protocol to assess micrographs from K2 or K3 cameras.
Contributors:
Cryoef
Available methods:
-
orientation analysis
- Protocol for analysing the orientation distribution of single-particle EM data. Find more information at http://www.mrc-lmb.cam.ac.uk/crusso/cryoEF/
Contributors:
Cryomethods
Plugin url https://github.com/mcgill-femr/scipion-em-cryomethods.
Available in Linux. Scientific software, Image processing, cryo emAvailable methods:
-
2D auto classifier (2D classification)
- Description missing, might be a new development.
-
3D auto classifier (3D classification)
- Description missing, might be a new development.
-
directional_pruning
- Analyze 2D classes as assigned to the different directions
-
volume selector
- Protocol to obtain a better initial volume using as input a set of volumes and particles. The protocol uses a small subset (usually 1000/2000) particles per classfrom the input set of particles to estimate a better and reliable volume(s) to use as initial volume in an automatic way.
Contributors:
 Cryosparc2
Cryosparc2
Available methods:
-
2D classification (2D classification)
- Wrapper to CryoSparc 2D clustering program. Classify particles into multiple 2D classes to facilitate stack cleaning and removal of junk particles. Also useful as a sanity check to investigate particle quality.
-
3D Classification
- 3D Classification (BETA) is a new job in cryoSPARC v3.3+ to analyze discrete heterogeneity in single particle cryo-EM datasets. This job currently implements a version of 3D classification without alignment — a classification routine that can complement the existing Heterogeneous Refinement job in finding new discrete classes of data.
-
3D Heterogeneous Refinement (3D classification)
- Heterogeneous Refinement simultaneously classifies particles and refines structures from n initial structures, usually obtained following an Ab-Initio Reconstruction. This facilitates the ability to look for small differences between structures which may not be obvious at low resolutions, and also to re-classify particles to aid in sorting.
-
3D helical refinement
- Reconstruct and refine a homogeneous helical assembly, with or without imposition and refinement of symmetry parameters. Helical Refinement (BETA) uses an algorithm that is conceptually similar to Egelman's Iterative Helical Real Space Reconstruction (IHRSR) algorithm, while incorporating the same maximum likelihood framework, accelerated branch-and-bound alignment algorithm, and optional Non-Uniform regularization as used in other cryoSPARC refinement jobs.
-
3D homogeneous refinement
- Protocol to refine a 3D map using cryosparc. Rapidly refine a single homogeneous structure to high-resolution and validate using the gold-standard FSC. Using new faster GPU code, and support for higher-order aberration (beam tilt, spherical aberration, trefoil, tetrafoil) correction and per-particle defocus refinement on the fly.
-
3D homogeneous refinement(Legacy) (3D refinement)
- Protocol to refine a 3D map using cryosparc. Rapidly refine a single homogeneous structure to high-resolution and validate using the gold-standard FSC.
-
3D non-uniform refinement
- Apply non-uniform refinement to achieve higher resolution and map quality, especially for membrane proteins. Non-uniform refinement iteratively accounts for regions of a structure that have disordered or flexible density causing local loss of resolution. Accounting for these regions and dynamically estimating their locations can significantly improve resolution in other regions as well as overall map quality by impacting the alignment of particles and reducing the tendency for refinement algorithms to over-fit disordered regions.
-
3D non-uniform refinement(Legacy) (3D refinement)
- Apply non-uniform refinement to achieve higher resolution and map quality, especially for membrane proteins. Non-uniform refinement iteratively accounts for regions of a structure that have disordered or flexible density causing local loss of resolution. Accounting for these regions and dynamically estimating their locations can significantly improve resolution in other regions as well as overall map quality by impacting the alignment of particles and reducing the tendency for refinement algorithms to over-fit disordered regions.
-
global ctf refinement
- Wrapper protocol for the Cryosparc's per-particle Global CTF refinement. Performs per-exposure-group CTF parameter refinement of higher-order aberrations, against a given 3D reference
-
homogeneous reconstruction
- Create a 3D reconstruction from input particles that already have alignments in 3D.
-
initial model (Initial model)
- Generate a 3D initial model _de novo_ from 2D particles using CryoSparc Stochastic Gradient Descent (SGD) algorithm.
-
local ctf refinement
- Wrapper protocol for the Cryosparc's per-particle Local CTF refinement. Performs per-particle defocus estimation for each particle in a dataset, against a given 3D reference structure.
-
local refinement
- Signal subtraction protocol of cryoSPARC. Subtract projections of a masked volume from particles.
-
naive local refinement(Legacy)
- Signal subtraction protocol of cryoSPARC. Subtract projections of a masked volume from particles.
-
sharppening
- Wrapper protocol for the Cryosparc's to calculate the sharpened map.
-
subtract projection
- Signal subtraction protocol of cryoSPARC. Subtract projections of a masked volume from particles.
-
symmetry expansion
- Duplicate particles around a point-group symmetry.
Contributors:

 Deepfinder
Deepfinder
Available methods:
-
Load Training Model
- Use two data-independent reconstructed tomograms to train a 3D cryo-CARE network.
-
annotate
- This protocol allows you to annotate macromolecules in your tomograms, using a visual tool.
-
cluster
- This protocol analyses segmentation maps and outputs particle coordinates and class.
-
display volume
- This protocol allows you to explore tomograms or segmentation maps with ortho-slices. The seegmentation map can be superimposed to the tomogram. Useful for visualising your results.
-
generate sphere target
- This protocol generates segmentation maps from annotations. These segmentation maps will be used as targets to train DeepFinder
-
import coordinates (Import)
- Protocol to import a DeepFinder object list as a set of 3D coordinates in Scipion
-
segment
- This protocol segments tomograms, using a trained neural network.
-
train
- This protocol launches the training procedure
Contributors:

 Dynamo
Dynamo
Available methods:
-
Subtomogram alignment
- This protocol will align subtomograms using Dynamo MRA Subtomogram Averaging
-
bin tomograms
- Reduce the size of a SetOfTomograms by a binning factor
-
coords to model
- Convert a SetOfCoordinates3D to a SetOfMeshes formatted to be appropiate to work with Dynamo protocols
-
import subtomos from Dynamo (Import)
- This protocol imports subtomograms with metadata generated by a Dynamo table. A Dynamo catalogue can be also imported in order to relate subtomograms with their original tomograms.
-
import tomograms from Dynamo (Import)
- This protocols imports a series of Tomogram stored in a Dynamo catalogue into Scipion. In order to avoid handling a MatLab binary, the script relies on MatLab itself to turn a binary MatLab object into an Structure which can be afterwards read by Python.
-
model manager
- Model manger from Dynamo for Mesh creation. After opening the desired Tomogram, a surface model will be created and loaded in Dynamo automatically. Once the points of the Mesh have been defined, close Dynamo to save automatically your data. If a dialog asking to keep the models loaded in memory appears, click on 'Keep it!!' or close the dialog.
-
model workflow
- Apply a model workflow to a SetOfMeshes generated by Dynamo Boxing protocol. This workflow will use the models created by the user to create the corresponding cropping meshes needed to extract the crop points
-
subBoxing
- SubBoxing using Dynamo
-
vectorial extraction
- Extraction of subtomograms using Dynamo
-
vectorial picking
- Manual vectorial picker from Dynamo. After choosing the Tomogram to be picked, the tomo slicer from Dynamo will be direclty loaded with all the models previously saved in the disk (if any). This picking will only save the "user points" defined in a set of models. It is possible to create several models at once in a given tomogram. Once the coordinates are defined, the models are automatically saved in the catalogue and registered. Currently the following Dynamo models are supported: - Ellipsoidal Vesicle
Contributors:

ESRF
ISPyB monitor for facility ESRF
Plugin url https://github.com/scipion-em/scipion-em-esrf.
Available in Linux. Scientific software, Image processing, cryo emAvailable methods:
-
monitor to ISPyB at the ESRF
- Monitor to communicated with ISPyB system at ESRF.
Contributors:

 Eman2
Eman2
Available methods:
-
boxer (Particle picking)
- Semi-automated particle picker for SPA. Uses EMAN2 e2boxer.py.
-
boxer auto (Particle picking)
- Automated particle picker for SPA. Uses EMAN2 (versions 2.2+) e2boxer.py
-
ctf auto (CTF estimation)
- This protocol wraps *e2ctf_auto.py* EMAN2 program. It automates the CTF fitting and structure factor generation process.
-
initial model (Initial model)
- This protocol wraps *e2initialmodel.py* EMAN2 program. It will take a set of class-averages/projections and build a set of 3-D models suitable for use as initial models in single particle reconstruction. The output set is theoretically sorted in order of quality (best one is numbered 1), though it's best to look at the other answers as well. See more details in: http://blake.bcm.edu/emanwiki/EMAN2/Programs/e2initialmodel
-
initial model SGD (Initial model)
- This protocol wraps *e2initialmodel_sgd.py* EMAN2 program. This program makes initial models using a (kind of) stochastic gradient descent approach. It is recommended that the box size of particles is around 100.
-
reconstruct (3D reconstruction)
- This protocol wraps *e2make3d.py* EMAN2 program. Reconstructs 3D volumes using a set of 2D images. Euler angles are extracted from the 2D image headers and symmetry is imposed. Several reconstruction methods are available. The fourier method is the default and recommended reconstructor.
-
refine 2D (2D classification)
- This protocol wraps *e2refine2d.py* EMAN2 program. This program is used to produce reference-free class averages from a population of mixed, unaligned particle images. These averages can be used to generate initial models or assess the structural variability of the data. They are not normally themselves used as part of the single particle reconstruction refinement process, which uses the raw particles in a reference-based classification approach. However, with a good structure, projections of the final 3-D model should be consistent with the results of this reference-free analysis. This program uses a fully automated iterative alignment/MSA approach. You should normally target a minimum of 10-20 particles per class-average, though more is fine. Default parameters should give a good start, but are likely not optimal for any given system. Note that it does have the --parallel option, but a few steps of the iterative process are not parallellised, so don't be surprised if multiple cores are not always active.
-
refine 2D bispec (2D classification)
- This protocol wraps *e2refine2d_bispec.py* EMAN2 program. This program is used to produce reference-free class averages from a population of mixed, unaligned particle images. These averages can be used to generate initial models or assess the structural variability of the data. They are not normally themselves used as part of the single particle reconstruction refinement process, which uses the raw particles in a reference-based classification approach. However, with a good structure, projections of the final 3-D model should be consistent with the results of this reference-free analysis. This variant of the program uses rotational/translational invariants derived from the bispectrum of each particle.
-
refine easy (3D refinement)
- This protocol wraps *e2refine_easy.py* EMAN2 program.This is the primary single particle refinement program in EMAN2.1+.It replaces earlier programs such as e2refine.py and e2refine_evenodd.py.Major features of this program: * While a range of command-line options still exist. You should not normally specify more than a few basic requirements. The rest will be auto-selected for you. * This program will split your data in half and automatically refine the halves independently to produce a gold standard resolution curve for every step in the refinement. * An HTML report file will be generated as this program runs, telling you exactly what it decided to do and why, as well as giving information about runtime, etc while the job is still running. * The gold standard FSC also permits us to automatically filter the structure at each refinement step. The resolution you specify is a target, NOT the filter resolution.
-
sparx gaussian picker (Particle picking)
- Automated particle picker for SPA. Uses Sparx gaussian picker. For more information see http://sparx-em.org/sparxwiki/e2boxer
-
tilt validate
- This protocol wraps the *e2tiltvalidate.py* EMAN2 program. It performs tilt validation using the method described in Rosenthal and Henderson, JMB (2003).
Contributors:





 Emfacilities
Emfacilities
This plugin allows to use different utils for cryo-EM facilities (like monitors) within the Scipion framework.
Plugin url https://github.com/scipion-em/scipion-em-facilities.
Available in Linux. Scientific software, Image processing, cryo emAvailable methods:
-
2d streamer
- This protocol will monitor an input set of particles (usually in streaming) and will run/schedule many copies of a given 2D classification protocol but using subsets of the input particles as the 2D classification input.
-
Track used items
- This protocol will track the items (micrographs, classes2D,...) that has been used in a scipion protocol to generate a final volume. If the ids have been maintained, it will also track the not used items.
-
ctf monitor
- check CPU, mem and IO usage.
-
monitor summary
- Provide some summary of the basic steps of the Scipion-Box: - Import movies - Align movies (global and/or local) - CTF estimation - Movie gain estimation.
-
movie gain monitor
- check CPU, mem and IO usage.
-
system_monitor
- check CPU, mem and IO usage.
Contributors:
 Empiar
Empiar
This project is a Scipion plugin to make depositions to https://www.ebi.ac.uk/pdbe/emdb/empiar
Plugin url https://github.com/scipion-em/scipion-em-empiar.
Available in Linux. Scientific software, Image processing, cryo emAvailable methods:
-
empiar downloader (Import)
- Downloads movies from EMPIAR and registers them.
Contributors:
 Emxlib
Emxlib
Available methods:
-
emx export (Export)
- Export micrographs, coordinates or particles to EMX format. EMX is a joint initiative for data exchange format between different EM software packages.
Contributors:
 Fsc3d
Fsc3d
Available methods:
-
estimate resolution (Local resolution)
- Protocol to calculate 3D FSC. 3D FSC is software tool for quantifying directional resolution using 3D Fourier shell correlation volumes. Find more information at https://github.com/nysbc/Anisotropy
Contributors:
Gautomatch
Available methods:
-
auto-picking (Particle picking)
- Automated particle picker for SPA. Gautomatch is a GPU accelerated program for accurate, fast, flexible and fully automatic particle picking from cryo-EM micrographs with or without templates.
Contributors:
 Grigoriefflab
Grigoriefflab
DEPRECATED! Plugin to use Grigorieff Lab (not CisTEM) programs within the Scipion framework.
Plugin url https://github.com/scipion-em/scipion-em-grigoriefflab.
Available in Linux. Scientific software, Image processing, cryo emAvailable methods:
-
ctffind4 (CTF estimation)
- Estimates CTF on a set of micrographs using either ctffind3 or ctffind4 program. To find more information about ctffind4 go to: http://grigoriefflab.janelia.org/ctffind4
-
ctftilt (CTF estimation)
- Estimates CTF on a set of tilted micrographs using ctftilt program.
-
frealign
- Protocol to refine a 3D map using Frealign. The algorithms implementedare optimized to perform efficiently the correction for the contrasttransfer function of the microscope and refinement of three-dimensionalreconstructions.
-
frealign classify (3D classification)
- Protocol to classify 3D using Frealign. Frealign employsmaximum likelihood classification for single particle electroncryo-microscopy.Particle alignment parameters are determined by maximizing ajoint likelihood that can include hierarchical priors, whileclassification is performed by expectation maximization of amarginal likelihood.
-
mag distortion correct
- This program automatically corrects anisotropic magnification distortion using previously estimated parameters. It works on a set of movies.
-
mag distortion correct (coords)
- This program automatically corrects anisotropic magnification distortion using previously estimated parameters. It works on a set of coordinates.
-
mag distortion estimate
- This program automatically estimates anisotropic magnification distortion from a set of images of a standard gold shadowed diffraction grating
-
summovie (Movie alignment)
- Summovie generates frame sums that can be used in subsequent image processing steps and optionally applies an exposure-dependent filter to maximize the signal at all resolutions in the frame averages.
-
unblur (Movie alignment)
- Unblur is used to align the frames of movies recorded on an electron microscope to reduce image blurring due to beam-induced motion.
Contributors:



Imagic
This plugin includes two protocols to provide wrappers around Multivariate Statistical Analysis (MSA) module of IMAGIC software suite
Plugin url https://github.com/scipion-em/scipion-em-imagic.
Available in Linux. Scientific software, Image processing, cryo emAvailable methods:
-
msa (2D classification)
- This protocols wraps MSA-RUN program of IMAGIC. It calculates eigenimages (eigenvectors) and eigenvalues of a set of input aligned images using an iterative eigenvector algorithm optimized for (extremely) large data sets.
-
msa-classify (2D classification)
- This protocols wraps MSA-CLASSIFY program of IMAGIC. It is based on variance-oriented hierarchical ascendant classification program (an enhanced Ward-type algorithm).
Contributors:
Imod
Available methods:
-
Apply transformation
- Compute the interpolated tilt-series from its transform matrix. More info: https://bio3d.colorado.edu/imod/doc/man/newstack.html
-
Automatic CTF estimation
- CTF estimation of a set of input tilt-series using the IMOD procedure. More info: https://bio3d.colorado.edu/imod/doc/man/ctfplotter.html
-
CTF correction
- CTF correction of a set of input tilt-series using the IMOD procedure. More info: https://bio3d.colorado.edu/imod/doc/man/ctfphaseflip.html
-
CTF estimation (CTF estimation)
- CTF estimation of a set of input tilt-series using the IMOD procedure. More info: https://bio3d.colorado.edu/imod/doc/man/ctfplotter.html
-
Coarse prealignment
- Tilt-series cross correlation alignment based on the IMOD procedure. More info: https://bio3d.colorado.edu/imod/doc/man/tiltxcorr.html
-
Dose filter
- Tilt-series dose filtering based on the IMOD procedure. More info: https://bio3d.colorado.edu/imod/doc/man/mtffilter.html
-
Etomo interactive
- Simple wrapper around etomo to manually reconstruct a Tomogram. More info: https://bio3d.colorado.edu/imod/doc/etomoTutorial.html
-
Exclude views
- excludeviews - Reversibly remove views from a tilt series stack If you use this protocol, make sure tis output tilt series is use for everything else CTF estimation, per particle per tilt, tomogram reconstruction.... More info: https://bio3d.colorado.edu/imod/doc/man/excludeviews.html
-
Fiducial alignment
- Construction of a fiducial model and alignment of tilt-series based on the IMOD procedure. More info: https://bio3d.colorado.edu/imod/doc/man/tiltalign.html https://bio3d.colorado.edu/imod/doc/man/model2point.html https://bio3d.colorado.edu/imod/doc/man/imodtrans.html https://bio3d.colorado.edu/imod/doc/man/newstack.html https://bio3d.colorado.edu/imod/doc/man/ccderaser.html
-
Generate fiducial model
- Construction of a fiducial model and alignment of tilt-series based on the IMOD procedure. More info: https://bio3d.colorado.edu/imod/doc/man/autofidseed.html https://bio3d.colorado.edu/imod/doc/man/beadtrack.html https://bio3d.colorado.edu/imod/doc/man/model2point.html
-
Gold bead picker 3D
- 3-dimensional gold bead picker using the IMOD procedure. More info: https://bio3d.colorado.edu/imod/doc/man/findbeads3d.html
-
Import tomo CTFs (Import)
- Protocol to import estimations of CTF series from tilt-series into Scipion.
-
Import transformation matrix (Import)
- Import the transformation matrices assigned to an input set of tilt-series
-
Manual CTF estimation
- CTF estimation of a set of input tilt-series using the IMOD procedure. Runs the protocol through the interactive GUI. The resulting defocus values MUST BE SAVED manually by the user. More info: https://bio3d.colorado.edu/imod/doc/man/ctfplotter.html
-
Tilt-series preprocess
- Normalize input tilt-series and change its storing formatting. More info: https://bio3d.colorado.edu/imod/doc/man/newstack.html
-
Tomo preprocess
- Normalize input tomogram and change its storing formatting. More info: https://bio3D.colorado.edu/imod/doc/newstack.html https://bio3D.colorado.edu/imod/doc/binvol.html
-
Tomo projection
- Re-project a tomogram given a geometric description (axis and angles). More info: https://bio3d.colorado.edu/imod/doc/man/xyzproj.html
-
Tomo reconstruction
- Tomogram reconstruction procedure based on the IMOD procedure. More info: https://bio3d.colorado.edu/imod/doc/man/tilt.html
-
X-rays eraser
- Erase X-rays from aligned tilt-series based on the IMOD procedure. More info: https://bio3d.colorado.edu/imod/doc/man/ccderaser.html
Contributors:
Legacy
A "bag" for protocols that has been deprecated
Available in Linux. Scientific software, Image processing, cryo emAvailable methods:
-
unknown protocol
- Protocols that were renamed, deprecated or not found when loading scipion. Opening a project without some plugins will turn some protocols into legacy protocols.
Contributors:
 Localrec
Localrec
Available methods:
-
Set origin to subvolume
- Set the origin and sampling values assigned to a 3D map so that the subvolume fits the original, larger volume
-
define subparticles (3D refinement)
- This class contains a re-implementation to a method for the localized three-dimensional reconstruction of such subunits. After determining the particle orientations, local areas corresponding to the subunits can be extracted and treated as single particles.
-
extract subparticles
- Extract computed sub-particles from a SetOfParticles.
-
filter subparticles
- This protocol mainly filters output particles from two protocols: extend symmetry and localized subparticles. It can filter the particles (sub-particles) according to spatial distance, view, and angular distance.
-
particles subset by subparticles
- This protocol make a subset of particles for which there is at least one sub-particle.
-
stitch subvolumes
- Generate a full volume from a sub-volume applying a point group symmetry operation. An example of usage is to generate the adenovirus capsid from its asymmetric unit.
Contributors:

 Locscale
Locscale
Available methods:
-
local sharpening (Local resolution)
- This protocol computes contrast-enhanced cryo-EM maps by local amplitude scaling using a reference model.
Contributors:
Motioncorr
Available methods:
-
align tilt-series movies (Movie alignment)
- This protocol wraps motioncor2 movie alignment program developed at UCSF. Motioncor2 performs anisotropic drift correction and dose weighting (written by Shawn Zheng @ David Agard lab)
-
movie alignment (Movie alignment)
- This protocol wraps motioncor2 movie alignment program developed at UCSF. Motioncor2 performs anisotropic drift correction and dose weighting (written by Shawn Zheng @ David Agard lab)
Contributors:

NovaCtf
Available methods:
-
tomo ctf defocus (CTF estimation)
- Defocus estimation of each tilt-image procedure based on the novaCTF procedure. More info: https://github.com/turonova/novaCTF
-
tomo ctf reconstruction (3D reconstruction)
- Tomogram reconstruction with ctf correction procedure based on the novaCTF procedure. More info: https://github.com/turonova/novaCTF
-
tomo ctf reconstruction
- Tomogram reconstruction and ctf correction procedure based on the novaCTF procedure. More info: https://github.com/turonova/novaCTF
Contributors:
 Phenix
Phenix
Available methods:
-
dock and rebuild predicted model
- Rebuild Docked Predicted Model. Rebuild predicted model morphs and rebuilds a model produced by AlphaFold, RoseTTAFold and other prediction software into a cryo EM map, using a set of docked domains from the predicted model as a template.
-
dock in map
- Docking of a PDB (one or several copies) into a map
-
dock predicted model
- Dock Predicted Model. Dock predicted model docks the domains from a model produced by AlphaFold, RoseTTAFold and other prediction software into a cryo EM map. It uses the connectivity of the model as a restraint in the docking process so that the docked domains normally are in a reasonable arrangement. It can take map symmetry into account.
-
emringer
- EMRinger is a Phenix application to validate the agreement betweenthe initial map and the derived low-resolution atomic structure. This programsamples the density around Chi1 angles of protein sidechains. Electronicdensity and appropriate rotameric angles must coincide for each residue ifthe atomic structure backbone has been perfectly fitted to the map.
-
molprobity
- MolProbity is a Phenix application to validate the geometry of anatomic structure inferred from an electron density map.
-
process predicted model
- Process Predicted Model Replace values in b-factor field with estimated B values. Optionally remove low-confidence residues and split into domains.
-
real space refine
- Tool for extensive real-space refinement of an atomic structure against the map provided. The map can be derived from X-ray or neutron crystallography, or cryoEM. The program obtains a model that fits the map as well as possible having appropriate geometry. The model should not show validation outliers, such as Ramachandran plot or rotamer outliers.
-
rebuild predicted model
- Rebuild Docked Predicted Model. Rebuild predicted model morphs and rebuilds a model produced by AlphaFold, RoseTTAFold and other prediction software into a cryo EM map, using a set of docked domains from the predicted model as a template.
-
refinementBase
- MolProbity is a Phenix application to validate the geometry of anatomic structure derived from a cryo-EM density map.
-
search fit
- given a chain of n alanines, a 3D map and a sequence search for the subsequence of n aminoacids that better fits in the density. Only works if the atomic structure has a single chain
-
superpose pdbs
- Superpose two PDBs so that they optimally match
-
validation_cryoem
- MolProbity is a Phenix application to validate the geometry of anatomic structure inferred from an electron density map.
Contributors:
Pkpd
Plugin url https://github.com/cossorzano/scipion-pkpd.
Available in Linux. Scientific software, Image processing, cryo emAvailable methods:
-
BE power analysis
- Power analysis for Bioequivalence studies. For further help, see https://cran.r-project.org/web/packages/PowerTOST/vignettes/PA.html https://cran.r-project.org/web/packages/PowerTOST/PowerTOST.pdf https://cran.r-project.org/web/packages/PowerTOST/index.html Protocol created by http://www.kinestatpharma.com
-
Exp1 SubGr2 Mean
- Compare two means from two subgroups from the same experiment . Protocol created by http://www.kinestatpharma.com
-
Exp2 SubGr2 Kolmogorov
- Check if two distributions come from the same distribution using the Kolmogorov Smirnov test. Protocol created by http://www.kinestatpharma.com
-
Exp2 SubGr2 Mean
- Compare two means from two subgroups from the same experiment . Protocol created by http://www.kinestatpharma.com
-
Mahalanobis
- Experiment 1 defines the mean and covariance for the Mahalanobis distance. Then, the Mahalanobis distance of all elements in Experiment 1 with respect to the mean is calculated If a second experiment is given, then all distances from the second to the mean of the first experiment are also calculated. Protocol created by http://www.kinestatpharma.com
-
ODE bootstrap
- Bootstrap of an ODE protocol
-
ODE refinement
- Refinement of an ODE protocol. The parameters are reestimated with a finer sampling rate.
-
PK simulate
- Simulate a population of ODE parameters. These parameters can be specifically given, from a bootstrap population, a previous fitting, or an experiment. AUC0t and AUMC0t are referred to each dose (that is, t=0 is the beginning of the dose). Ctau is the concentration at the end of the dose. Tmin, Tmax, and Ttau are referred to the beginning of the dose. This protocol writes an Excel file (nca.xlsx) in which all the simulations and doses are written.
-
PK simulate (complex)
- Simulate a complex sequence of doses. Each dose has its own PK profile, and the overall profile is the addition of all the individual profiles. Only the parameters from the first fitting of each model are considered. The first dose goes with the first model, the second dose with the second model, ... A single profile is simulated that is the addition of the different simulations for each one of the input models.
-
absorption rate
- Estimation of the absorption rate for a non-intravenous route. The estimation is performed after estimating the elimination rate. The experiment is determined by the Protocol created by http://www.kinestatpharma.com. See the theory at http://www.pharmpress.com/files/docs/Basic%20Pharmacokinetics%20sample.pdf
-
allometric scaling
- Compute the allometric scaling between several spicies. This fits a model of the form Y=k*X^a where X can be any variable (although weight is normally used). The protocol can also leave out some parameters of the input model, and for these parameters, a simple average is calculated.
-
apply allometric
- Apply an allometric scaling previously calculated to an incoming experiment. The labels specified by the allometric scaling model will be rescaled to the target weight. Note that depending on the exponent of the fitting you may want to use a different predictor (weight*maximum lifespan potential, or weight*brain weight) see the rule of exponents (Mahmood and Balian 1996).
-
average sample
- Produce an experiment with a single sample whose value is the average of all the input samples. Protocol created by http://www.kinestatpharma.com
-
bioavailability nca
- Estimate bioavailability as F=(AUCpo/Dpo) / (AUCiv/Div) [Gabrielsson 2010, p. 546], i.e., the ratio between the oral and intravenous AUC normalized by their respective doses. Protocol created by http://www.kinestatpharma.com
-
change units
- Change units of a given variable. Protocol created by http://www.kinestatpharma.com
-
change via
- Change via of administration This protocol may also be used to change the bioavailability or the tlag Protocol created by http://www.kinestatpharma.com
-
compare experiments
- This protocol compares two experiments by plotting a summary of both. Protocol created by http://www.kinestatpharma.com
-
create experiment
- Create experiment. Protocol created by http://www.kinestatpharma.com
-
create experiment
- Create experiment. Protocol created by http://www.kinestatpharma.com
-
create label
- Create label by performing calculations on already existing labels. Protocol created by http://www.kinestatpharma.com
-
create label 2 Exps
- Create label by performing calculations on already existing labels from two different experiments. The protocol assumes that the same samples are present in both experiments and calculations are performed only on those samples with the same name in both experiments. Protocol created by http://www.kinestatpharma.com
-
cumulated dose
- Create label with the cumulated dose between two time points. Protocol created by http://www.kinestatpharma.com
-
deconvolution Loo-Riegelman
- Calculate the absorption profile of an in vivo concentration profile using the Loo-Riegelman approach. This is only valid for profiles that have been modelled with a two-compartments PK model. The formula is Fabs(t)=(Cp(t)+Cperipheral(t)+K10*AUC0t(t))/(K10*AUC0inf) where K10=Cl/V and Cperipheral(t_n)=k12*Delta Cp*Delta t/2+k12/k21 * Cp(t_n-1)(1-exp(-k21*Delta t))+Cperipheral(t_n-1)*exp(-k21*Delta t) In this implementation it is assumed that AUC0inf is the last AUC0t observed, meaning that Cp(t) has almost vanished in the last samples. This protocol is much more accurate when the input Cp(t) is reinterpolated to a small time step like 0.5 minutes. Reference: Leon Shargel, Susanna Wu-Pong, Andrew B.C. Yu. Applied Biopharmaceutics & Pharmacokinetics, 6e. McGraw Hill, 1999. Chap. 7
-
deconvolution Wagner Nelson
- Calculate the absorption profile of an in vivo concentration profile using the Wagner-Nelson approach. This is only valid for profiles that have been modelled with a monocompartment PK model. The formula is Fabs(t)=(Cp(t)+Ke*AUC0t(t))/(Ke*AUC0inf) where Ke=Cl/V In this implementation it is assumed that AUC0inf is the last AUC0t observed, meaning that Cp(t) has almost vanished in the last samples
-
deposition pattern
- Read deposition parameters associated to some substance See Hartung2020. Protocol created by http://www.kinestatpharma.com
-
dissol Levy
- Calculate the Levy plot between two dissolution experiments. Each experiment may have several profiles and all vs all profiles are calculated
-
dissol deconv
- Deconvolve the drug dissolution from a compartmental model.
-
dissol deconv Fourier
- Deconvolve the drug dissolution from a compartmental model. It does the deconvolution in Fourier so that it only uses the impulse response of the compartmental model. This impulse response only depends on the distribution, metabolism and excretion (DME) part of the ADME properties, meaning that it overcomes the limitations of a poor modelling of the raise of the concentration. On the other side, it has the disadvantage of considering the noise as true fluctuations due to the absorption.
-
dissol f1 and f2
- Calculate the f1 and f2 from two dissolution profiles. The bootstrap confidence interval is bias corrected and accelarated.
-
dissol ivivc
- Calculate the in vitro-in vivo correlation between two experiments. Each experiment may have several profiles and all vs all profiles are calculated. You may scale the time between the two sets of experiments
-
dissol ivivc generic
- Calculate the in vitro-in vivo correlation between two experiments. Each experiment may have several profiles and all vs all profiles are calculated. You may scale the time between the two sets of experiments
-
dissol ivivc join avg
- Join several IVIVCs into a single one. The strategy is to compute the average of all the plots involved in the IVIVC process: 1) tvivo -> tvitro; 2) tvitro -> Adissol; 3) Adissol->FabsPredicted. The plot tvivo-Fabs comes after the IVIVC process, while the plot tvivo-FabsOrig is the observed one in the input files. These two plots need not be exactly the same.
-
dissol ivivc join recalculate
- Join several IVIVCs into a single one. Look for a single time transformation and response transformation such that all input pairs of in vitro and in vivo profiles are satisfied simultaneously.
-
dissol ivivc splines
- Calculate the in vitro-in vivo correlation between two experiments. Each experiment may have several profiles and all vs all profiles are calculated. You may scale the time between the two sets of experiments
-
dissol levyplot join avg
- Join several Levy plots into a single one. The strategy is to compute the average of all the plots involved in the Levy plot process: 1) tvivo -> tvitro
-
dissol target
- Given an in-vivo absorption profile (assumed to be between 0 and 100), and the IVIVC parameters specified in this protocol, the protocol calculates the target in-vitro dissolution profile
-
dose escalation
- Given a set of binary responses (toxicity, response/not response, ...), estimate the next dose for a target response Protocol created by http://www.kinestatpharma.com
-
drop measurements
- Filter measurements. Protocol created by http://www.kinestatpharma.com
-
elimination rate
- Fit a single exponential to the input data. Protocol created by http://www.kinestatpharma.com
-
export to csv (Export)
- Export experiment to CSV. Protocol created by http://www.kinestatpharma.com
-
filter measurements
- Filter measurements. Protocol created by http://www.kinestatpharma.com
-
filter population
- Filter a population by some criterion Protocol created by http://www.kinestatpharma.com
-
filter samples
- Filter samples. Protocol created by http://www.kinestatpharma.com
-
fit bootstrap
- Bootstrap estimate of generic fit models. Protocol created by http://www.kinestatpharma.com
-
fit dissolution
- Fit a dissolution model. The observed measurement is modelled as Y=f(t).Confidence intervals calculated by this fitting may be pessimistic because it assumes that all model parametersare independent, which are not. Use Bootstrap estimates instead. Protocol created by http://www.kinestatpharma.com
-
fit exponentials
- Fit a set of exponentials. The observed measurement is modelled as Y=sum_{i=1}^N c_i exp(-lambda_i * X).Confidence intervals calculated by this fitting may be pessimistic because it assumes that all model parametersare independent, which are not. Use Bootstrap estimates instead. Protocol created by http://www.kinestatpharma.com
-
fit pd generic
- Fit a generic model. The observed measurement is modelled as Y=f(X).Confidence intervals calculated by this fitting may be pessimistic because it assumes that all model parametersare independent, which are not. Use Bootstrap estimates instead. Protocol created by http://www.kinestatpharma.com
-
gather fitting
- Gather several fittings coming from a split experiment into a single fitting. Protocol created by http://www.kinestatpharma.com
-
import experiment (Import)
- Protocol to import an PKPD experiment Protocol created by http://www.kinestatpharma.com
-
import from csv
- Import experiment from CSV. You may use WebPlotDigitizer (https://apps.automeris.io/wpd) to generate this CSV, or Excel. Protocol created by http://www.kinestatpharma.com
-
import from excel (Import)
- Import experiment from Excel. Protocol created by http://www.kinestatpharma.com
-
import from winnonlin (Import)
- Import experiment from Winnonlin. Protocol created by http://www.kinestatpharma.com
-
inverse Loo-Riegelman
- Given a profile of amount absorbed, find the central and peripheral concentrations that gave raise to it. This is an inverse Loo-Riegelman problem. The parameters of this protocol are defined as microconstants. Remind that k10=Cl*V, k12=Clp*V, k21=Clp*Vp
-
iv two-compartments
- Fit a two-compartmentx model with intravenous absorption to a set of measurements (any arbitrary dosing regimen is allowed) The central compartment is referred to as C, while the peripheral compartment as Cp. The differential equation is V dC/dt = -(Cl+Clp) * C + Clp * Cp + dD/dt, Vp dCp/dt = Clp * C - Clp * Cp where C is the concentration of the central compartment, Cl the clearance, V and Vp the distribution volume of the central and peripheral compartment, Clp is the distribution rate between the central and the peripheral compartments, and D the input dosing regime. Confidence intervals calculated by this fitting may be pessimistic because it assumes that all model parameters are independent, which are not. Use Bootstrap estimates instead. Protocol created by http://www.kinestatpharma.com
-
ivivc internal validity
- This protocol compares the AUC and Cmax predicted from an in vitro-in vivo experiment and the AUC and Cmax of the in vivo population from which the IVIV correlation was estimated. There should not be differences larger than 15%% between the two sets of variables. Protocol created by http://www.kinestatpharma.com
-
join samples
- Join samples. Vias are not prefixed. If they are repeated, those from Experiment 1 prevail. Protocol created by http://www.kinestatpharma.com
-
lung parameters
- Produce a description of the lung physiological parameters See Hartung2020. Protocol created by http://www.kinestatpharma.com
-
merge labels
- Merge the labels of Experiment 2 into the samples of Experiment 1. If a label is in Experiment 1 and in Experiment 2, it remains the value from Experiment 1. Protocol created by http://www.kinestatpharma.com
-
merge populations
- Merge two populations. Both populations must have the same labels Protocol created by http://www.kinestatpharma.com
-
monocompartment urine
- Fit a monocompartmental model to a set of plasma and urine (cumulated) measurements ((any arbitrary dosing regimen is allowed) The differential equation is dC/dt = -Cl * C/V + 1/V * dD/dt and dA/dt = fe * Cl * C where C is the concentration, Cl the clearance, V the distribution volume, D the input dosing regime and fe the fraction excreted.Confidence intervals calculated by this fitting may be pessimistic because it assumes that all model parametersare independent, which are not. Use Bootstrap estimates instead. Protocol created by http://www.kinestatpharma.com
-
nca ev
- Non-compartmental analysis of a non-intravenous bolus. Protocol created by http://www.kinestatpharma.com
-
nca iv exponentials
- Non-compartmental analysis based on an exponential fitting. Protocol created by http://www.kinestatpharma.com
-
nca iv observations
- Non-compartmental analysis based on observations. Protocol created by http://www.kinestatpharma.com
-
nca numeric
- Non-compartmental analysis just based on the samples. The results are valid only up to T. It is valid for any kind of via (intravenous, extravascular, ...). Protocol created by http://www.kinestatpharma.com
-
ode two vias
- Simultaneous fit of data obtained by different vias, e.g. IV and PO, but it can be any two vias and any two dosing regimes, dissolution profiles, etc. It is supposed that the PK model in both cases is the same (e.g. two monocompartments, two two-compartments, ...
-
one-compartment linkpd
- Fit a mono-compartment model to a set of plasma and effect measurements (any arbitrary dosing regimen is allowed) The differential equation is dC/dt = -Cl * C/V -Clp *(C-Cp)/V + 1/V * dD/dt, dCb/dt=Cl*C/Vb+Clb*(C-Cb)/Vp and E=E0+a*Cb^b/(Cbm^b+Cb^b) where C is the concentration, Cl the total clearance (metabolic and excretion), V the distribution volume, Clb is the Clearance to the biophase compartment, Vb is the volume of the biophase compartment, D the input dosing regime E is the measured effect, E0 a baseline effect, a and b fitting constants and Cbm the biophase concentration at which half the maximum effect is attained.Confidence intervals calculated by this fitting may be pessimistic because it assumes that all model parametersare independent, which are not. Use Bootstrap estimates instead. Protocol created by http://www.kinestatpharma.com
-
one-compartment pd
- Fit a mono-compartment model to a set of plasma and effect measurements (any arbitrary dosing regimen is allowed) The differential equation is dC/dt = -Cl * C/V + 1/V * dD/dt and E=E0+a*C^b/(Cm^b+C^b) where C is the concentration, Cl the total clearance (metabolic and excretion), V the distribution volume, D the input dosing regime E is the measured effect, E0 a baseline effect, a and b fitting constants and Cm the biophase concentration at which half the maximum effect is attained.Confidence intervals calculated by this fitting may be pessimistic because it assumes that all model parametersare independent, which are not. Use Bootstrap estimates instead. Protocol created by http://www.kinestatpharma.com
-
operate experiment
- Create a new label or measurement to an existing experiment. Protocol created by http://www.kinestatpharma.com
-
particle size distribution
- Estimate the particle size distribution parameters. The size is supposed to be log-normal and the mean and standard deviation of the log-normal distribution are estimated. Given a set of particle sizes {x1, x2, ..., xN} and a set of bin occupancies (they can be in absolute or relative counts or mass) {p1, p2, ..., pN}, this protocol estimates the parameters of the log-normal distribution that is compatible with these observations. It also reports the fitting quality of the distribution. If it is descending order, then it is assumed that p1=Prob{x1<=x<x2}, p2=Prob{x2<=x<x3}, ..., pN=Prob{x<=xN} If it is ascending order, then it is assumed that p1=Prob{x<=x1}, p2=Prob{x1<x<=x2}, ..., pN=Prob{xN<=x} The program calculates the mu and sigma of the log-normal distribution and their exponentials.
-
pk monocompartment
- Fit a monocompartmental model to a set of measurements obtained by oral doses (any arbitrary dosing regimen is allowed) The differential equation is dC/dt = -Cl * C/V + 1/V * dD/dt where C is the concentration, Cl the clearance, V the distribution volume, and D the input dosing regime.Confidence intervals calculated by this fitting may be pessimistic because it assumes that all model parametersare independent, which are not. Use Bootstrap estimates instead. Protocol created by http://www.kinestatpharma.com
-
pk monocompartment conv
- Fit a monocompartmental model to a set of measurements obtained by oral doses (any arbitrary dosing regimen is allowed) The differential equation is dC/dt = -Cl * C/V + 1/V * dD/dt where C is the concentration, Cl the clearance, V the distribution volume, and D the input dosing regime.Confidence intervals calculated by this fitting may be pessimistic because it assumes that all model parametersare independent, which are not. Use Bootstrap estimates instead. The forward model is implemented by convolution instead of by numerical solution of the differential equation. Protocol created by http://www.kinestatpharma.com
-
pk monocompartment intrinsic
- Fit a monocompartmental model to a set of measurements obtained by oral doses (any arbitrary dosing regimen is allowed) The differential equation is dC/dt = -Clint * C/V + 1/V * dD/dt and Clint=Vmax/(Km+C) where C is the concentration, Clint the intrinsic clearance, V the distribution volume, Vmax is the maximum processing capability of the metabolic pathway degrading the drug, Km is the Michaelis-Menten constant, and D the input dosing regime. Confidence intervals calculated by this fitting may be pessimistic because it assumes that all model parameters are independent, which are not. Use Bootstrap estimates instead. Protocol created by http://www.kinestatpharma.com
-
pk three-compartments
- Fit a three-compartments model to a set of measurements (any arbitrary dosing regimen is allowed) The central compartment is referred to as C, while the peripheral compartments as Cpa and Cpb. A and B peripheral compartments are both connected to the central compartment. The differential equation is V dC/dt = -(Cl+Clpa+Clpb) * C + Clpa * Cpa + Clpb * Cpb + dD/dt, Vpa dCpa/dt = Clpa * C - Clpa * Cpa Vpb dCpb/dt = Clpb * C - Clpb * Cpb where C is the concentration of the central compartment, Cl the clearance, V and Vpa, Vpb the distribution volume of the central and peripheral compartments, Clpa and Clpb are the distribution rates between the central and the peripheral compartments, and D the input dosing regime. Confidence intervals calculated by this fitting may be pessimistic because it assumes that all model parameters are independent, which are not. Use Bootstrap estimates instead. Protocol created by http://www.kinestatpharma.com
-
pk two-compartments
- Fit a two-compartments model to a set of measurements (any arbitrary dosing regimen is allowed) The central compartment is referred to as C, while the peripheral compartment as Cp. The differential equation is V dC/dt = -(Cl+Clp) * C + Clp * Cp + dD/dt, Vp dCp/dt = Clp * C - Clp * Cp where C is the concentration of the central compartment, Cl the clearance, V and Vp the distribution volume of the central and peripheral compartment, Clp is the distribution rate between the central and the peripheral compartments, and D the input dosing regime. Confidence intervals calculated by this fitting may be pessimistic because it assumes that all model parameters are independent, which are not. Use Bootstrap estimates instead. Protocol created by http://www.kinestatpharma.com
-
pk two-compartments autoinduction
- Fit a two-compartment model to a set of measurements (any arbitrary dosing regimen is allowed) The central compartment is referred to as C, while the peripheral compartment as Cp. The differential equation is V dC/dt = -(Cl+Clp) * C + Clp * Cp + dD/dt, Vp dCp/dt = Clp * C - Clp * Cp being Cl=Cl0-a*Cp where C is the concentration of the central compartment, Cl0 the basal clearance, V and Vp the distribution volume of the central and peripheral compartment, Clp is the distribution rate between the central and the peripheral compartments, and D the input dosing regime. As the concentration in the peripheral compartment increases, the clearance is slowed. Confidence intervals calculated by this fitting may be pessimistic because it assumes that all model parameters are independent, which are not. Use Bootstrap estimates instead. Protocol created by http://www.kinestatpharma.com
-
pk two-compartments conv
- Fit a two-compartmentx model to a set of measurements (any arbitrary dosing regimen is allowed) The central compartment is referred to as C, while the peripheral compartment as Cp. The differential equation is V dC/dt = -(Cl+Clp) * C + Clp * Cp + dD/dt, Vp dCp/dt = Clp * C - Clp * Cp where C is the concentration of the central compartment, Cl the clearance, V and Vp the distribution volume of the central and peripheral compartment, Clp is the distribution rate between the central and the peripheral compartments, and D the input dosing regime. Confidence intervals calculated by this fitting may be pessimistic because it assumes that all model parameters are independent, which are not. Use Bootstrap estimates instead. The forward model is implemented by convolution instead of by numerical solution of the differential equation. Protocol created by http://www.kinestatpharma.com
-
pk two-compartments intrinsic
- Fit a two-compartmentx model to a set of measurements (any arbitrary dosing regimen is allowed) The central compartment is referred to as C, while the peripheral compartment as Cp. The differential equation is V dC/dt = -(Clint+Clp) * C + Clp * Cp + dD/dt, Vp dCp/dt = Clp * C - Clp * Cp and Clint=Vmax/(Km+C) where C is the concentration of the central compartment, Cl the clearance, V and Vp the distribution volume of the central and peripheral compartment, Clp is the distribution rate between the central and the peripheral compartments, Vmax is the maximum processing capability of the metabolic pathway degrading the drug, Km is the Michaelis-Menten constant,and D the input dosing regime. Note that the intrinsic clearance occurs at the central volume. Confidence intervals calculated by this fitting may be pessimistic because it assumes that all model parameters are independent, which are not. Use Bootstrap estimates instead. Protocol created by http://www.kinestatpharma.com
-
pk two-compartments intrinsic+Cl
- Fit a two-compartmentx model to a set of measurements (any arbitrary dosing regimen is allowed) The central compartment is referred to as C, while the peripheral compartment as Cp. The differential equation is V dC/dt = -(Clint+Clp+Cl) * C + Clp * Cp + dD/dt, Vp dCp/dt = Clp * C - Clp * Cp and Clint=Vmax/(Km+C) where C is the concentration of the central compartment, Cl the clearance, V and Vp the distribution volume of the central and peripheral compartment, Clint is a saturated clearance Clp is the distribution rate between the central and the peripheral compartments, Vmax is the maximum processing capability of the metabolic pathway degrading the drug, Km is the Michaelis-Menten constant,and D the input dosing regime. Note that the intrinsic clearance occurs at the central volume. Confidence intervals calculated by this fitting may be pessimistic because it assumes that all model parameters are independent, which are not. Use Bootstrap estimates instead. Protocol created by http://www.kinestatpharma.com
-
pk two-compartments intrinsic, metabolite
- Fit a two-compartmentx model to a set of measurements (any arbitrary dosing regimen is allowed) The central compartment is referred to as C, while the peripheral compartment as Cp. The differential equation is V dC/dt = -(Clint+Clp) * C + Clp * Cp + dD/dt, Vp dCp/dt = Clp * C - Clp * Cp, dCm/dt=Clint*C/Vm-Clm*Cm/Vm and Clint=Vmax/(Km+C) where C is the concentration of the central compartment, V, Vp, Vm the distribution volume of the central, peripheral and metabolite compartment, Clp is the distribution rate between the central and the peripheral compartments, Clm is the clearance of metabolite at the metabolite compartments, Vmax is the maximum processing capability of the metabolic pathway degrading the drug into the metabolite, Km is the Michaelis-Menten constant,and D the input dosing regime. Note that the intrinsic clearance occurs at the central volume. Confidence intervals calculated by this fitting may be pessimistic because it assumes that all model parameters are independent, which are not. Use Bootstrap estimates instead. Protocol created by http://www.kinestatpharma.com
-
regression labels
- Perform a regression between two labels Protocol created by http://www.kinestatpharma.com
-
scale dose
- Scale to common dose If the system is linear, then we may scale all measurements as if all individuals had been given the same dose. In this way we may construct a cleaner version of the response (by averaging) and use this cleaner version to find the initial parameters of the rest of samples. This protocol calculates the total dose of each individual and constructs new individuals such that Cnew = Cold * newDose/oldDose The old dose is evaluated in a period (by default, 1 week) that should include all doses Protocol created by http://www.kinestatpharma.com
-
simulate PK response
- This protocol simulates the pharmacokinetic response of an ODE model when it is given a single dose of an drug whose release is modelled by an in vitro fitting and an in vitro-in vivo correlation.
-
simulate dissolution
- Simulate a dissolution profile Protocol created by http://www.kinestatpharma.com
-
simulate dose escalation
- Simulate a dose escalation Protocol created by http://www.kinestatpharma.com
-
simulate drug interactions
- Simulate drug interactions as recommended in EMA CHMP/EWP/560/95 Protocol created by http://www.kinestatpharma.com
-
simulate generic
- Simulate a generic pharmacodynamic response Y=f(X). Protocol created by http://www.kinestatpharma.com
-
simulate inhalation
- Simulate inhalation PK See Hartung2020. Protocol created by http://www.kinestatpharma.com
-
simulate liver flow
- Simulate the concentration of a compound (typically an enzyme inhibitor) at liver. Protocol created by http://www.kinestatpharma.com
-
split/gather experiment
- Split an experiment into small pieces, or gather these small pieces into an experiment. Protocol created by http://www.kinestatpharma.com
-
statistics labels
- Calculate statistics of the labels Protocol created by http://www.kinestatpharma.com
-
substance parameters
- Produce a description of the properties of a substance related to inhalation See Hartung2020. Protocol created by http://www.kinestatpharma.com
-
two-compartments both
- Fit a two-compartments model to a set of plasma and peripheral compartment measurements ((any arbitrary dosing regimen is allowed) The differential equation is dC/dt = -Cl * C/V -Clp *(C-Cp)/V + 1/V * dD/dt, dCp/dt=Cl*C/Vp+Clp*(C-Cp)/Vp where C is the concentration, Cl the total clearance (metabolic and excretion), V the distribution volume, Clp is the Clearance to the peripheric compartment, Vp is the volume of the peripheric compartment, and D the input dosing regime. This protocol assumes that you have measures of both the central and peripheral compartments.Confidence intervals calculated by this fitting may be pessimistic because it assumes that all model parametersare independent, which are not. Use Bootstrap estimates instead. Protocol created by http://www.kinestatpharma.com
-
two-compartments both pd
- Fit a mono-compartment model to a set of plasma and effect measurements (any arbitrary dosing regimen is allowed) The differential equation is dC/dt = -Cl * C/V -Clp *(C-Cp)/V + 1/V * dD/dt, dCp/dt=Cl*C/Vp+Clp*(C-Cp)/Vp and E=E0+a*C^b/(Cm^b+C^b) where C is the concentration, Cl the total clearance (metabolic and excretion), V the distribution volume, Clp is the Clearance to the peripheric compartment, Vp is the volume of the peripheric compartment, and D the input dosing regime. E is the measured effect, E0 a baseline effect, a and b fitting constants and Cm the biophase concentration at which half the maximum effect is attained. This protocol assumes that you have measures of both the central and peripheral compartments.Confidence intervals calculated by this fitting may be pessimistic because it assumes that all model parametersare independent, which are not. Use Bootstrap estimates instead. Protocol created by http://www.kinestatpharma.com
-
two-compartments urine
- Fit a two-compartments model to a set of plasma and urine (cumulated) measurements ((any arbitrary dosing regimen is allowed) The differential equation is dC/dt = -Cl * C/V -Clp *(C-Cp)/V + 1/V * dD/dt, dCp/dt=Cl*C/Vp+Clp*(C-Cp)/Vp and dA/dt = fe * Cl * C where C is the concentration, Cl the total clearance (metabolic and excretion), V the distribution volume, Clp is the Clearance to the peripheric compartment, Vp is the volume of the peripheric compartment, D the input dosing regime and fe the fraction excreted.Confidence intervals calculated by this fitting may be pessimistic because it assumes that all model parametersare independent, which are not. Use Bootstrap estimates instead. Protocol created by http://www.kinestatpharma.com
Contributors:
Powerfit_scipion
Program from Alexandre Bonvin and Gydo van Zundert (Utrecht University) for rigid body fitting of an atomic structure into a 3D volume
Plugin url https://github.com/scipion-em/scipion-em-powerfit.
Available in Linux. Scientific software, Image processing, cryo emAvailable methods:
-
powerfit
- Protocol for fitting a PDB into a 3D volume This is actually a wrapper to the program Powerfit. See documentation at: http://www.bonvinlab.org/education/powerfit
Contributors:
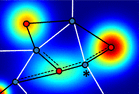 Pyseg
Pyseg
Available methods:
-
2D classification
- Unsupervised and deterministic classification of membrane-bound particles
-
fils
- filter a MbGraphMCF (Mean Cumulative Function) object by extracting a filament network
-
graphs
- analyze a GraphMCF (Mean Cumulative Function) from a segmented membrane
-
import star file PySeg (Import)
- This protocol imports subtomograms from a STAR file generated by PySeg
-
picking
- extract particles from a filament network of a oriented single membrane graph
-
posrec
- post-process already reconstructed particles: rot angle randomization and membrane suppression
-
preseg membranes
- Segment membranes into membranes, inner surroundings and outer surroundings
Contributors:
 Relion
Relion
Available methods:
-
2D class ranker
- Relion protocol to auto-select 2D class averages.
-
2D classification (2D classification)
- This protocol runs Relion 2D classification.
-
3D auto-refine (3D refinement)
- Protocol to refine a 3D map using Relion.Relion employs an empirical Bayesian approach to refinementof (multiple) 3D reconstructionsor 2D class averages in electron cryo-microscopy (cryo-EM). Manyparameters of a statistical model are learned from the data,whichleads to objective and high-quality results.
-
3D classification (3D classification)
- Protocol to classify 3D using Relion Bayesian approach. Relion employs an empirical Bayesian approach to refinement of (multiple) 3D reconstructions or 2D class averages in electron cryo-EM. Many parameters of a statistical model are learned from the data, which leads to objective and high-quality results.
-
3D initial model (Initial model)
- This protocols creates a 3D initial model using Relion. Generate a 3D initial model _de novo_ from 2D particles using Relion Stochastic Gradient Descent (SGD) algorithm.
-
3D multi-body (3D refinement)
- Relion protocol for multi-body refinement. This approach models flexible complexes as a user-defined number of rigid bodies that move independently of each other. Using separate focused refinements with iteratively improved partial signal subtraction, improved reconstructions are generated for each of the defined bodies. Moreover, using PCA on the relative orientations of the bodies over all particle images in the data set, we generate movies that describe the most important motions in the data.
-
assign optics groups
- Assign Optics Group name and related parameters to an input set. Input set can be: movies, micrographs or particles. Warning: all optics parameters from the input set itself are removed!
-
auto-picking (Particle picking)
- This protocol runs Relion autopicking (version > 3.0). This Relion protocol uses the 'relion_autopick' program to pick particles from micrographs, either using references (2D averages or 3D volumes) The wrapper implementation does not read/write any FOM maps compared to Relion
-
auto-picking LoG (Particle picking)
- This Relion protocol uses 'relion_autopick' program for the Laplacian of Gaussian (LoG) option.
-
bayesian polishing
- Wrapper protocol for the Relion's Bayesian Polishing. As of release 3.0, Relion also implements a new Bayesian approach to beam induced motion correction. This approach aims to optimise a regularised likelihood, which allows us to associate with each hypothetical set of particle trajectories a prior likelihood that favors spatially coherent and temporally smooth motion without imposing any hard constraints. The smoothness prior term requires three parameters that describe the statistics of the observed motion. To estimate the prior that yields the best motion tracks for this particular dataset, we can first run the program in 'training mode'. Once the estimates have been obtained, one can then run the program again to fit tracks for the motion of all particles in the data set and to produce adequately weighted averages of the aligned movie frames.
-
calculate fsc
- Relion protocol to calculate various FSC curves using relion_image_handler.
-
center averages
- Align class averages by their center of mass using *relion_image_handler*. (With *--shift_com* option)
-
clean project
- Run Relion gentle clean procedure for the whole project.
-
compress movies
- Using *relion_convert_to_tiff* to compress a set of movies.
-
create 3d mask (Masking)
- This protocols creates a 3D mask using Relion. The mask is created from a 3d volume or by comparing two input volumes.
-
crop/resize volumes
- This protocol rescales/resizes 3D volumes using relion_image_handler.
-
ctf refinement (3D refinement)
- Wrapper protocol for the Relion's CTF refinement.
-
estimate gain reference
- Using *relion_convert_to_tiff* to estimate the gain reference from a set of movies.
-
expand symmetry
- This protocols wraps relion_particle_symmetry_expand program. Given an input set of particles with angular assignment, expand the set by applying a pseudo-symmetry.
-
export coordinates (Export)
- Export coordinates from Relion to be used outside Scipion.
-
export ctf (Export)
- Export a SetOfCTF to a Relion STAR file.
-
export particles (Export)
- Export particles from Relion to be used outside Scipion.
-
import coordinates (Import)
- Import coordinates from a particles star file.
-
local resolution (Local resolution)
- This protocol does local resolution estimation using Relion. This program basically performs a series of post-processing operations with a small soft, spherical mask that is moved over the entire map, while using phase-randomisation to estimate the convolution effects of that mask.
-
motion correction (Movie alignment)
- Wrapper for the Relion's implementation of motioncor algorithm.
-
movie particles extraction
- Protocol to extract particles from a set of coordinates
-
particle polishing
- This protocols runs particle polishing using Relion. This Relion protocol tracks particle movement in movie frames (from previous movie refinement run), applies a resolution and dose-dependent weighting scheme for each frame and finally sums them together, producing so-called shiny, or polished particles.
-
particles extraction
- Protocol to extract particles using a set of coordinates.
-
post-processing
- Sharpen a 3D reference map and estimate the gold-standard FSC curves
-
preprocess particles
- This protocol wraps relion_preprocess program. It is used to perform normalisation, filtering or scaling of the particles.
-
reconstruct (3D reconstruction)
- This protocol reconstructs a volume using Relion. Reconstruct a volume from a given set of particles. The alignment parameters will be converted to a Relion star file and used as direction projections to reconstruct.
-
remove preferential views
- Protocol to remove preferential views from a particle set. Inspired by https://github.com/leschzinerlab/Relion
-
sort particles
- Relion particle sorting protocol. It calculates difference images between particles and their aligned (and CTF-convoluted) references, and produces Z-score on the characteristics of these difference images (such as mean, standard deviation, skewness, excess kurtosis and rotational symmetry).
-
subtract projection
- Signal subtraction protocol of Relion. Subtract volume projections from the experimental particles. The particles must have projection alignment in order to properly generate volume projections.
-
symmetrize volume
- Symmetrize a volume using Relion programs: *relion_align_symmetry* and *relion_image_handler*.
Contributors:



 RelionTomo
RelionTomo
Available methods:
-
3D Classification of subtomograms
- 3D Classification of subtomograms.
-
3D subtomogram auto-refine (3D refinement)
- Protocol to refine a 3D map using Relion. Relion employs an empiricalBayesian approach to refinement of (multiple) 3D reconstructionsor 2D class averages in electron cryo-microscopy (cryo-EM). Manyparameters of a statistical model are learned from the data,whichleads to objective and high-quality results.
-
3D subtomogram classification (3D classification)
- Protocol to classify 3D using Relion. Relion employs an empirical Bayesian approach to refinement of (multiple) 3D reconstructions or 2D class averages in electron cryo-microscopy (cryo-EM). Many parameters of a statistical model are learned from the data,which leads to objective and high-quality results.
-
Apply operation to Relion particles
- Operate on the particles star file
-
Auto-refinement of subtomograms
- Auto-refinement of subtomograms.
-
CTF 3D estimation (CTF estimation)
- Generates the CTF star and MRC files needed by relion for the CTF3D.
-
De novo 3D initial model
- Generate a de novo 3D initial model from the pseudo-subtomograms.
-
Make pseudo-subtomograms
- Make pseudo-subtomograms
-
Prepare data for Relion 4
- Prepare data for Relion 4
-
Rec. particle averaging subtomograms
- This protocol reconstructs a volume using Relion. Reconstruct a volume from a given set of particles. The alignment parameters will be converted to a Relion star file and used as direction projections to reconstruct.
-
Reconstruct particle from tilt series
- Reconstruct particle from the original tilt series images
-
Reconstruct tomograms from prepare data prot (3D reconstruction)
- This protocol reconstructs a single tomogram using Relion. It is very useful to check if the protocol "Prepare data" has been applied correctly (in terms of flip options, for example).
-
Tomo CTF refine
- Tomo CTF refine
-
Tomo frame align
- Tomo frame align
-
extract coordinates from pseudo-subtomograms
- Protocol to extract a set of 3D coordinates from a set of pseudo-subtomograms
-
import coordinates 3D from a star file (Import)
- Protocol to import a set of 3D coordinates from a star file
-
import subtomograms from a star file (Import)
- Protocol to import a set of subtomograms from a star file
-
tomo reconstruct (3D reconstruction)
- This protocol reconstructs a volume using Relion. Reconstruct a volume from a given set of particles. The alignment parameters will be converted to a Relion star file and used as direction projections to reconstruct.
Contributors:
 Resmap
Resmap
Available methods:
-
local resolution (Local resolution)
- ResMap is software tool for computing the local resolution of 3D density maps from electron cryo-microscopy (cryo-EM). Please find the manual at https://sourceforge.net/projects/resmap-latest
Contributors:
Sidesplitter
Available methods:
-
local filter (Local resolution)
- Protocol for mitigating local over-fitting by filtering. Find more information at https://github.com/StructuralBiology-ICLMedicine/SIDESPLITTER
Contributors:
 Simple
Simple
This plugin allows to use Simple programs within the Scipion framework.
Plugin url https://github.com/scipion-em/scipion-em-simple.git@python3_migration.
Available in Linux. Scientific software, Image processing, cryo emAvailable methods:
-
In development
- Description missing, might be a new development.
-
prime
- Produces one or several initial volumes using simple prime
Contributors:
 Sphire
Sphire
Available methods:
-
cryolo import (Import)
- Protocol to import an existing crYOLO training model. The model will be registered as an output of this protocol and it can be used later for further training or for picking.
-
cryolo picking (Particle picking)
- Picks particles in a set of micrographs either manually or in a supervised mode.
-
cryolo tomo picking (Particle picking)
- Picks particles in a set of micrographs either manually or in a supervised mode.
-
cryolo training (Particle picking)
- Picks particles in a set of micrographs either manually or in a supervised mode.
-
janni denoising
- Protocol to denoise a set of micrographs in the project.
Contributors:
 Spider
Spider
Available methods:
-
align ap sr (2D alignment)
- This protocol wraps SPIDER AP SR command. Reference-free alignment (both translational and rotational) of an image series. See detailed description at: [[http://spider.wadsworth.org/spider_doc/spider/docs/man/apsr.html][SPIDER's AP SR online manual]]
-
align pairwise (2D alignment)
- This protocol wraps SPIDER AP SR command (pairwise alignment). Reference-free alignment (both translational and rotational) of an image series. This alignment scheme aligns a pair of images at a time and then averages them. Then, the averages of each of those pairs are aligned and averaged, and then pairs of those pairs, etc. Compared to [[http://spider.wadsworth.org/spider_doc/spider/docs/man/apsr.html][AP SR]], this alignment scheme appears to be less random, which chooses seed images as alignment references. For more information, see Step 2b at [[http://spider.wadsworth.org/spider_doc/spider/docs/techs/MSA/index.html#pairwise][SPIDER's MDA online manual]]
-
ca pca (2D classification)
- Protocol for Correspondence Analysis (CA) or Principal Component Analysis (PCA). CA is the preferred method of finding inter-image variations. PCA computes the distance between data vectors with Euclidean distances, while CA uses Chi-squared distance. CA is superior because it ignores differences in exposure between images, eliminating the need to rescale between images. In contrast, PCA seems to be more robust: less likely to be trapped in an infinite loop of numerical inaccuracy. For more info see: [[http://spider.wadsworth.org/spider_doc/spider/docs/techs/classification/tutorial.html#CAPCA][SPIDER MDA documentation]]
-
classify diday (2D classification)
- This protocol wraps SPIDER CL CLA command. Performs automatic clustering using Diday's method and Hierarchical Ascendant Classification (HAC) using Ward's criterion on factors produced by CA or PCA.
-
classify kmeans (2D classification)
- This protocol wraps SPIDER CL KM command. Performs automatic K-Means clustering and classification on factors produced by CA or PCA.
-
classify ward (2D classification)
- This protocol wraps SPIDER CL HC command. Finds clusters of images/elements in factor space (or a selected subspace) by using Diday's method of moving centers, and applies hierarchical ascendant classification (HAC) (using Ward's method) to the resulting cluster centers.
-
create 2d mask (Masking)
- This protocol creates a 2D mask using SPIDER. In the step following this one, dimension-reduction, the covariance of the pixels in all images will be computed. Only pixels under a given mask will be analyzed. If this step is performed, a mask that follows closely the contour the particle of interest will be used. Absent a custom-made mask, a circular mask will be used. For non-globular structures, this customized mask will reduce computational demand and the likelihood of numerical inaccuracy in the next dimension-reduction step. On the other hand, given the power of modern computers, this step may be unnecessary.
-
filter particles
- Apply Fourier filters to an image or a volume using Spider FQ or FQ NP. To improve boundary quality the image is padded with the average value to twice the original size during filtration if padding is selected. See more documentation at: [[http://spider.wadsworth.org/spider_doc/spider/docs/man/fq.html][SPIDER's FQ online manual]]
-
reconstruct fourier (3D reconstruction)
- This protocol wraps SPIDER BP 32F command. Simple reconstruction protocol using Fourier back projection. Mainly used for testing conversion of Euler angles.
-
refine 3D (3D refinement)
- Reference-based refinement using SPIDER AP SHC and AP REF commands. Iterative refinement improves the accuracy in the determination of orientations. This improvement is accomplished by successive use of more finely-sampled reference projections. Two different workflows are suggested: with defocus groups or without (gold-standard refinement). For more information, see: [[http://spider.wadsworth.org/spider_doc/spider/docs/techs/recon/mr.html][SPIDER documentation on projection-matching]]
Contributors:
Susan
This plugin provides wrappers for Susan software: subtomogram averaging (StA) workflow for CryoET based on sub-stacks of images instead of sub-volumes of tomograms.
Plugin url https://github.com/scipion-em/scipion-em-susantomo.
Available in Linux. Scientific software, Image processing, cryo emAvailable methods:
-
average and reconstruct
- Description missing, might be a new development.
-
create a subset (Tools)
- Description missing, might be a new development.
-
ctf estimation (CTF estimation)
- Description missing, might be a new development.
-
multi-reference alignment
- Description missing, might be a new development.
Contributors:
 Tomo
Tomo
Available methods:
-
2D particles to subtomograms
- Protocol to create a set of subtomograms from a selected 2D particles.
-
2d coordinates to 3d coordinates
- Turns 2d coordinates into set of 3d coordinates. Works in coordination with 'tomograms to micrographs' protocol
-
Compose Tilt Serie
- Compose in streaming a set of tilt series based on a sets of micrographs and mdoc files. Three time parameters are abailable for the streaming behaviour: Time for next tilt, Time for next micrograph and Time for next Tilt Serie
-
Tilt-series assign alignment
- Assign the transformation matrices from an input set of tilt-series to a target one.
-
Tilt-series consensus alignment
- Perform a consensus of a set of alignments for the same tilt series. Returns the average alignment matrix of the consensus alignments and its standard deviation of shift and angle.
-
Tilt-series convert coords3D
- Scipion protocol to convert a set of tilt-series coordinates 3d to a set of coordinates 3d associated to a set of tomograms.
-
assign alignment
- Assign the alignment stored in a set of Subtomograms/Coordinates3D to another set. Both sets should have same pixel size (A/px). The Subtomograms/Coordinates3D with the alignment can also be a subset of a bigger set.
-
assign tomograms to tomo masks (segmentations)
- This protocol assign tomograms to tomomasks (segmentations).
-
assign tomos to subtomos
- This protocol assign tomograms to subtomograms that have been imported before without tomograms. Subtomograms should contain the name of the original tomogram in their own file name.
-
average tilt-series movies
- Simple protocol to average TiltSeries movies as basic motion correction. It is used mainly for testing purposes.
-
consensus classes subtomo
- Compare several SetOfClassesSubTomograms. Return the intersection of the input classes.
-
ctf validate
- Validate a set of CTF tomo series and separate into two sets (good and bad tomo series )
-
extract 3D coordinates
- Extract the coordinates information from a set of subtomograms. This protocol is useful when we want to re-extract the subtomograms (maybe resulting from classification) with the original dimensions. It can be also handy to visualize the resulting subtomograms in their location on the tomograms.
-
import 3D coordinates from scipion (Import)
- Protocol to import a set of 3d coordinates from Scipion sqlite file
-
import set of coordinates 3D (Import)
- Protocol to import a set of tomograms to the project
-
import subtomograms (Import)
- Protocol to import a set of tomograms to the project
-
import tilt-series (Import)
- Protocol to import tilt series.
-
import tilt-series movies (Import)
- Protocol to import tilt series movies.
-
import tomograms (Import)
- Protocol to import a set of tomograms to the project
-
import tomomasks (segmentations) (Import)
- Protocol to import a set of tomomasks (segmentations) to the project
-
split even/odd tomos/subtomos
- Protocol to split set of tomograms or subtomograms in even/odd sets by element id.
-
tomograms to micrographs
- Turns tomograms into set of micrographs to apply SPA picking methods.
Contributors:

Tomoj
Plugin url https://github.com/scipion-em/scipion-em-tomoj.
Available in Linux. Scientific software, Image processing, cryo emAvailable methods:
-
xcorr prealignment
- Tilt-series' cross correlation alignment based on the TomoJ procedure. More info: http://u759.sfbiophys.org/software/update/20140207/Manual_TomoJ_2.24.pdf
Contributors:
 TomosegmemTV
TomosegmemTV
Scipion plugin for segmentation and annotation of membranes in tomograms (with TomoSegmenTV and memb-annotator, respectively)
Plugin url https://github.com/scipion-em/scipion-em-tomosegmemtv.
Available in Linux. Scientific software, Image processing, cryo emAvailable methods:
-
Resize segmented or annotated volume (Tools)
- Resize segmented volumes or annotated (TomoMasks).
-
Tomogram segmentation (Masking)
- Segment membranes in tomogram
-
annotate segmented membranes (Masking)
- Manual annotation tool for segmented membranes
Contributors:
Tomotwin
This plugin provides a wrapper for TomoTwin software: Particle picking in Tomograms using triplet networks and metric learning
Plugin url https://github.com/scipion-em/scipion-em-tomotwin.
Available in Linux. Scientific software, Image processing, cryo emAvailable methods:
-
reference-based picking (Particle picking)
- Description missing, might be a new development.
Contributors:
 Topaz
Topaz
Available methods:
-
topaz import (Import)
- Protocol to import an existing topaz training model. The model will be registered as an output of this protocol and it can be used later for further training or for picking.
-
topaz picking
- Perform a picking using a topaz model
-
topaz training (Particle picking)
- Train and save a Topaz model
Contributors:

 Warp
Warp
Available methods:
-
deconvolve 2D
- Protocol to deconvolve (Wiener-like filter) a set of micrographs
-
deconvolve 3D
- Protocol to deconvolve (Wiener-like filter) a set of tomograms.
Contributors:
 Xmipp2
Xmipp2
Plugin url https://github.com/I2PC/scipion-em-xmipp2.git.
Available in Linux. Scientific software, Image processing, cryo emAvailable methods:
-
mltomo
- Protocol to align subtomograms using MLTomo. It only supports alignment on the axis Y. MLTomo aligns and classifies 3D images with missing data regions in Fourier space, e.g. subtomograms or RCT reconstructions, by a 3D multi-reference refinement based on a maximum-likelihood (ML) target function.
Contributors:
 Xmipp3
Xmipp3
Plugin to use Xmipp programs within the Scipion framework.
Plugin url https://github.com/i2pc/scipion-em-xmipp.
Available in Linux. Scientific software, Image processing, cryo emAvailable methods:
-
2D classes mapping
- Create a low dimensional mapping from a SetOfClasses2D with interactive selection of classes. Use mouse left-click to select/deselect classes individually or mouse right-click to select/deselect several classes.
-
2D kmeans clustering (2D classification)
- Classifies a set of particles using a clustering algorithm to subdivide the original dataset into a given number of classes.
-
3d bionotes
- Protocol for checking annotations This is actually a wrapper to 3D Bionotes. See documentation at: http://3dbionotes.cnb.csic.es
-
FlexAlign (Movie alignment)
- Wrapper protocol to Xmipp Movie Alignment by cross-correlation
-
XmippProtDeepConsSubSet
- Create subsets from the GUI for the Deep Consensus protocol. This protocol will be executed mainly calling the script 'pw_create_image_subsets.py' from the ShowJ gui. The enabled/disabled changes will be stored in a temporary sqlite file that will be read to create the new subset.
-
add noise particles
- Given a set of particles, the protocol will add noise to them The types of noise are Uniform, Student and Gaussian.
-
add noise volume/s
- Given a set of volumes, or a volume the protocol will add noise to them The types of noise are Uniform, Student and Gaussian.
-
align volume
- Aligns a set of volumes using cross correlation or a Fast Fourier method.
-
align volume and particles
- Aligns a volume (inputVolume) using a Fast Fourier method with respect to a reference one (inputReference). The obtained alignment parameters are used to align the set of particles (inputParticles) that generated the input volume.
-
align volume web
- Aligns a set of volumes using cross correlation. Based on Xmipp protocol for aligning volumes, but the parameters are restricted for ease of use.
-
align with cl2d
- Aligns a set of particles using the CL2D algorithm.
-
analyze local defocus
- Assigns to each micrograph a coefficient (R2) which evaluates the result of the local defocus adjustment and displays the local defocus for all the particles in each micrograph.
-
angular align - Zernike3D
- Protocol for flexible angular alignment based on Zernike3D basis.
-
angular graph consistency
- Performs soft alignment validation of a set of particles previously aligned confronting them using Graph filtered correlations representation. This protocol produces an histogram with two groups of particles.
-
apply 2d mask (Masking)
- Apply mask to a set of particles
-
apply 3d mask (Masking)
- Apply mask to a volume
-
apply alignment 2d
- Apply alignment parameters and produce a new set of images.
-
apply deformation field - Zernike3D
- Protocol for PDB deformation based on Zernike3D basis.
-
apply transformation matrix
- Apply transformation matrix of an aligned volume on a set of particles to modify their angular assignment. Note: These particles are practically related to the aligned volume (but before alignment).
-
assign tiltpairs
- From two sets of points (tilted and untilted) the protocol determines the affine transformation between these sets.
-
auto-picking (step 2) (Particle picking)
- Protocol to pick particles automatically in a set of micrographs using previous training
-
boost particles
- This protocol tries to boost the frequencies of the particles to imporve them, based on an adjustment on its correspondent projections from a reference volume.
-
break symmetry
- Given an input set of particles with angular assignment, find an equivalent angular assignment for a given symmetry. Be aware that input symmetry values follows Xmipp conventions as described in: http://xmipp.cnb.csic.es/twiki/bin/view/Xmipp/Symmetry
-
calculate strain
- Compare two states of a volume to analyze the local strains and rotations
-
center particles (Particle picking)
- Realignment of un-centered particles.
-
cl2d (2D classification)
- Classifies a set of images using a clustering algorithm to subdivide the original dataset into a given number of classes.
-
compare angles
- Compare two sets of angles. The output is a list of all common particles with the angular difference between both assignments. The output is constructed by keeping the information from the Set 1 and adding the shiftDiff and angularDiff.
-
compare reprojections
- Compares a set of classes or averages with the corresponding projections of a reference volume. The set of images must have a 3D angular assignment and the protocol computes the residues (the difference between the experimental images and the reprojections). The zscore of the mean and variance of the residues are computed. Large values of these scores may indicate outliers. The protocol also analyze the covariance matrix of the residual and computes the logarithm of its determinant [Cherian2013]. The extremes of this score (called zScoreResCov), that is values particularly low or high, may indicate outliers.
-
consensus clustering 3D (3D classification)
- Compare several SetOfClasses3D. Return the consensus clustering based on a objective function that uses the similarity between clusters intersections and the entropy of the clustering formed.
-
consensus local defocus
- This protocol compares the estimations of local defocus computed by different protocols for a set of particles
-
convert a PDB
- Convert a PDB file into a volume.
-
convert to pseudoatoms
- Converts an EM volume into pseudoatoms
-
core analysis
- Analyzes the core of a 2D classification. The core is calculated through the Mahalanobis distance from each image to the center of the class.
-
create 2d mask (Masking)
- Create a 2D mask. The mask can be created with a given geometrical shape (Circle, Rectangle, Crown...) or it can be obtained from operating on a 2d image or a previuous mask.
-
create 3d mask (Masking)
- Create a 3D mask. The mask can be created with a given geometrical shape (Sphere, Box, Cylinder...) or it can be obtained from operating on a 3d volume or a previous mask.
-
create gallery
- Create a gallery of projections from a volume. This gallery of projections may help to understand the images observed in the microscope.
-
crop/resize particles (Tools)
- Crop or resize a set of particles
-
crop/resize volumes (Tools)
- Crop or resize a set of volumes
-
ctf consensus (CTF estimation)
- Protocol to make a selection of meaningful CTFs in basis of the defocus values, the astigmatism, the resolution, other Xmipp parameters, and the agreement with a secondary CTF for the same set of micrographs.
-
ctf estimation (CTF estimation)
- Protocol to estimate CTF on a set of micrographs using Xmipp.
-
ctf_correct_wiener2d
- Perform CTF correction by Wiener filtering.
-
deep consensus picking
- Protocol to compute a smart consensus between different particle picking algorithms. The protocol takes several Sets of Coordinates calculated by different programs and/or different parameter settings. Let's say: we consider N independent pickings. Then, a neural network is trained using different subset of picked and not picked cooridantes. Finally, a coordinate is considered to be a correct particle according to the neural network predictions. In streaming, the network is trained and used to predict in batches. The network is trained until the number of particles set is reached, meanwhile, a preliminary output is generated. Once the threshold is reached, the final output is produced by batches.
-
deep denoising
- Description missing, might be a new development.
-
deep hand
- Protocol to returns handedness of structure from trained deep learning model
-
deep micrograph cleaner
- Protocol to remove coordinates in carbon zones or large impurities
-
deepEMhancer
- Given a map the protocol performs automatic deep post-processing to enhance visualization. Usage guide at https://github.com/rsanchezgarc/deepEMhancer
-
defocus group (CTF estimation)
- Given a set of CTFs group them by defocus value. The output is a metadata file containing a list of defocus values that delimite each defocus group.
-
deform pdb - Zernike3D
- Protocol for PDB deformation based on Zernike3D basis.
-
denoise particles
- Remove particles noise by filtering them. This filtering process is based on a projection over a basis created from some averages (extracted from classes). This filtering is not intended for processing particles. The huge filtering they will be passed through is known to remove part of the signal with the noise. However this is a good method for clearly see which particle are we going to process before it's done.
-
directional resolution MonoDir
- Given a map the protocol assigns local resolutions to each voxel of the map.
-
eliminate empty classes (2D classification)
- Takes a set of classes (or averages) and using statistical methods (variances of sub-parts of input image) eliminates those samples, where there is no object/particle (only noise is presented there). Threshold parameter can be used for fine-tuning the algorithm for type of data. Also discards classes with less population than a given percentage.
-
eliminate empty particles
- Takes a set of particles and using statistical methods (variance of variances of sub-parts of input image) eliminates those samples, where there is no object/particle (only noise is presented there). Threshold parameter can be used for fine-tuning the algorithm for type of data.
-
enrich
- Method to get two volume from different classes (with different conformation) and correcting (deforming) all images of one of the volumes (input volume) with respect to the another one as a reference, using optical flow algorithm. The output is a setOfParticles contaied deformed reference particles.
-
estimate local defocus (CTF estimation)
- Compares a set of particles with the corresponding projections of a reference volume. The set of particles must have a 3D angular assignment. This protocol refines the CTF, computing local defocus change. The maximun allowed defocus is a parameter introduced by the user (advanced). The protocol gives back the input set of particles with the refine local defocus and the defocus change with relation to the global defocus.
-
extract asymmetric unit (Export)
- generates files for volumes and FSCs to submit structures to EMDB
-
extract movie particles (Particle picking)
- Extract a set of Particles from each frame of a set of Movies.
-
extract particle pairs
- Protocol to extract particles from a set of tilted pairs coordinates
-
extract particles (Particle picking)
- Protocol to extract particles from a set of coordinates
-
filter particles
- Apply Fourier filters to a set of particles
-
filter volumes
- Apply Fourier filters to a set of volumes
-
generate reprojections
- Compares a set of classes or averages with the corresponding projections of a reference volume. The set of images must have a 3D angular assignment and the protocol computes the residues (the difference between the experimental images and the reprojections). The zscore of the mean and variance of the residues are computed. Large values of these scores may indicate outliers. The protocol also analyze the covariance matrix of the residual and computes the logarithm of its determinant [Cherian2013]. The extremes of this score (called zScoreResCov), that is values particularly low or high, may indicate outliers.
-
gl2d (2D classification)
- 2D alignment using Xmipp GPU Correlation algorithm.
-
gl2d static
- 2D alignment in semi streaming using Xmipp GPU Correlation. A previous set of classes must be provided to include the new images in the corresponding class although the representatives will be maintained.
-
gl2d streaming (2D classification)
- 2D alignment in full streaming using Xmipp GPU Correlation. The set of classes will be growing whilst new particle images are received.
-
helical symmetry
- Estimate helical parameters and symmetrize. Helical symmetry is defined as V(r,rot,z)=V(r,rot+k*DeltaRot,z+k*Deltaz).
-
highres (3D refinement)
- This is a 3D refinement protocol whose main input is a volume and a set of particles. The set of particles has to be at full size (the finer sampling rate available), but the rest of inputs (reference volume and masks) can be at any downsampling factor. The protocol scales the input images and volumes to a reasonable size depending on the resolution of the previous iteration. The protocol works with any input volume, whichever its resolution, as long as it is a reasonable initial volume for the set of particles. The protocol does not resolve the heterogeneous problem (it assumes an homogeneous population), although it is somewhat tolerant through the use of particle weights in the reconstruction process. It is recommended to perform several global alignment iterations before entering into the local iterations. The switch from global to local should be performed when a substantial percentage of the particles do not move from one iteration to the next. The algorithm reports the cross correlation (global alignment) or cost (local) function per defocus group, so that we can see which was the percentile of each particle in its defocus group. You may want to perform iterations one by one, and remove from one iteration to the next, those particles that worse fit the model.
-
kerdensom (2D classification)
- Classifies a set of images using Kohonen's Self-Organizing Feature Maps (SOM) and Fuzzy c-means clustering technique (FCM) . The kerdenSOM algorithm anneals from an initial high regularization factor to a final lower one, in a user-defined number of steps. KerdenSOM is an excellent tool for classification, especially when using a large number of data and classes and when the transition between the classes is almost continuous, with no clear separation between them. The input images must be previously aligned.
-
local MonoRes (Local resolution)
- Given a map the protocol assigns local resolutions to each voxel of the map.
-
local deepRes
- Given a map the protocol assigns local resolutions to each voxel of the map.
-
local resolution/local bfactor
- Given a local resolution map and an atomic model, this protocols provides the matching between the local resolution with the local bfactor per residue.
-
localdeblur sharpening (3D refinement)
- Given a resolution map the protocol calculate the sharpened map.
-
manual-picking (step 1) (Particle picking)
- Picks particles in a set of micrographs either manually or in a supervised mode.
-
metaprotocol golden highres
- Metaprotocol to run golden version of highres
-
metaprotocol heterogeneity
- Metaprotocol to run together all the protocols to discover discrete heterogeneity in a set of particles
-
metaprotocol heterogeneity output
- Metaprotocol to run together all the protocols to discover discrete heterogeneity in a set of particles
-
metaprotocol heterogeneity subset (Tools)
- Metaprotocol to select a set of particles from a 3DClasses and a Volume from a SetOfVolumes
-
ml2d (2D classification)
- Perform (multi-reference) 2D-alignment using a maximum-likelihood ( *ML* ) target function. Initial references can be generated from random subsets of the experimental images or can be provided by the user (this can introduce bias). The output of the protocol consists of the refined 2D classes (weighted averages over all experimental images). The experimental images are not altered at all. Although the calculations can be rather time-consuming (especially for many, large experimental images and a large number of references we strongly recommend to let the calculations converge.
-
mltomo
- Align and classify 3D images with missing data regions in Fourier space, e.g. subtomograms or RCT reconstructions, by a 3D multi-reference refinement based on a maximum-likelihood (ML) target function. See http://xmipp.cnb.csic.es/twiki/bin/view/Xmipp/Ml_tomo_v31 for further documentation
-
movie average (Movie alignment)
- Protocol to average movies
-
movie gain
- Estimate the gain image of a camera, directly analyzing one of its movies. It can correct the orientation of an external gain image (by comparing it with the estimated). Finally, it estimates the residual gain (the gain of the movie after correcting with a gain). The gain used in the correction will be preferably the external gain, but can also be the estimated gain if the first is not found. The same criteria is used for assigning the gain to the output movies (external corrected > external > estimated)
-
movie maxshift (Movie alignment)
- Protocol to make an automatic rejection of those movies whose frames move more than a given threshold. Rejection criteria: - *by frame*: Rejects movies with drifts between frames bigger than a certain maximum. - *by whole movie*: Rejects movies with a total travel bigger than a certain maximum. - *by frame and movie*: Rejects movies if both conditions above are met. - *by frame or movie*: Rejects movies if one of the conditions above are met.
-
movie resize
- Resize a set of movies. Only downsampling is allowed.
-
multiple fscs
- Compute the FSCs between a reference volume and a set of input volumes. A mask can be provided and the volumes are aligned by default.
-
multireference alignability
- Performs soft alignment validation of a set of particles confronting them against a given 3DEM map. This protocol produces particle alignment precision and accuracy parameters.
-
nma alignment
- Protocol for flexible angular alignment.
-
nma analysis
- Flexible angular alignment using normal modes
-
nma cluster
- Protocol executed when a cluster is created from NMA images and theirs deformations.
-
nma dimred
- This protocol will take the images with NMA deformations as points in a N-dimensional space (where N is the number of computed normal modes) and will project them in a reduced spaced (usually with less dimensions).
-
normalize strain
- Normalize the local strain and rotations amongst several runs
-
operate particles
- Apply an operation to two sets of particles
-
operate volumes
- Apply an operation to two sets of volumes
-
optical alignment (Movie alignment)
- Wrapper protocol to Xmipp Movie Alignment by Optical Flow
-
optical alignment (Movie alignment)
- Wrapper protocol to Xmipp Movie Alignment by Optical Flow
-
particle boxsize (Particle picking)
- Given a set of micrographs, the protocol estimate the particle box size.
-
phantom volume
- Create phantom volume from a feature description file using xmipp_phantom_create
-
pick noise (Particle picking)
- Protocol to pick noise particles
-
picking consensus (Particle picking)
- Protocol to estimate the agreement between different particle picking algorithms. The protocol takes several Sets of Coordinates calculated by different programs and/or different parameter settings. Let's say: we consider N independent pickings. Then, a coordinate is considered to be a correct particle if M pickers have selected the same particle (within a radius in pixels specified in the form). If you want to be very strict, then set M=N; that is, a coordinate represents a particle if it has been selected by all particles (this is the default behaviour). Then you may relax this condition by setting M=N-1, N-2, ... If you want to be very flexible, set M=1, in this way it suffices that 1 picker has selected the coordinate to be considered as a particle. Note that in this way, the cleaning of the dataset has to be performed by other means (screen particles, 2D and 3D classification, ...).
-
preprocess micrographs
- Protocol to preprocess a set of micrographs in the project. You can crop borders, remove bad pixels, etc.
-
preprocess particles
- Preprocess a set of particles. You can remove dust, normalize, apply threshold, etc
-
preprocess volumes
- Protocol for Xmipp-based preprocess for volumes
-
projection matching (3D refinement)
- 3D reconstruction and classification using multireference projection matching
-
random conical tilt
- Creates initial volumes by using a set of projections/classes from a tilted-pair picking process and using RCT algorithm.
-
ransac (Initial model)
- Computes an initial 3d model from a set of projections/classes using RANSAC algorithm. This method is based on an initial non-lineal dimensionality reduction approach which allows to select representative small sets of class average images capturing the most of the structural information of the particle under study. These reduced sets are then used to generate volumes from random orientation assignments. The best volume is determined from these guesses using a random sample consensus (RANSAC) approach.
-
reconstruct fourier (3D reconstruction)
- Reconstruct a volume using Xmipp_reconstruct_fourier from a given set of particles. The alignment parameters will be converted to a Xmipp xmd file and used as direction projections to reconstruct.
-
reconstruct significant (Initial model)
- This algorithm addresses the initial volume problem in SPA by setting it in a Weighted Least Squares framework and calculating the weights through a statistical approach based on the cumulative density function of different image similarity measures.
-
remove duplicates (Particle picking)
- This protocol removes coordinates that are closer than a given threshold. The remaining coordinate is the average of the previous ones.
-
resolution 3D
- Computes resolution by several methods
-
resolution fso
- Given two half maps the protocol estimates Fourier Shell Occupancy to determine the global anisotropy of the map. See more information here: https://github.com/I2PC/xmipp/wiki/FSO---Fourier-Shell-Occupancy
-
rotate volume
- Rotate a volume around x,y,z
-
rotational spectra
- Protocol to compute the rotational spectrum of the given particles.
-
rotational symmetry
- Estimate the orientation of a rotational axis and symmetrize. The user should know the order of the axis (two-fold, three-fold, ...) If this is unknown you may try several and see the most consistent results.
-
screen deep learning
- Protocol for screening particles using deep learning.
-
screen particles
- Protocol to attach different merit values to every particle metadata for subsequent pruning the set.There are different merit values to be calculated: - zScore evaluates the similarity of a particles with an average (lower zScore -> higher similarity). - SSNR evaluates the signal/noise ration in the Fourier space. - Variance evaluates the varaince on the micrographs context where the particle was picked.
-
shift particles
- This protocol shifts particles to center them into a point selected in a volume. To do so, it generates new shifted images and modify the transformation matrix according to the shift performed.
-
shift volume
- This protocol shifts a volume according to the input shifts
-
significant heterogeneity
- 3D Reconstruction with heterogeneous datasets
-
simulate ctf (CTF estimation)
- Simulate the effect of the CTF (no amplitude decay). A random defocus is chosen between the lower and upper defocus for each projection.
-
solid angles
- Construct image groups based on the angular assignment. All images assigned within a solid angle are assigned to a class. Classes are not exclusive and an image may be assigned to multiple classes
-
sph angular align
- Protocol for flexible angular alignment based on spherical harmonics.
-
sph struct map
- Protocol for structure mapping based on spherical harmonics.
-
sph volume deform
- Protocol for volume deformation based on spherical harmonics.
-
split frames
- Wrapper protocol to Xmipp split Odd Even
-
split volume
- Split volume in two
-
split volume hierarchical
- Construct image groups based on the angular assignment. All images assigned within a solid angle are assigned to a class. Classes are not exclusive and an image may be assigned to multiple classes
-
struct map
- Protocol for structure mapping based on correlation distance.
-
struct map - Zernike3D
- Protocol for structure mapping based on Zernike3D.
-
structure mapping
- A quantitive analysis of dissimilarities (distances) among the EM maps that placing the entire set of density maps in to a common space of comparison.The approach is based on statistical analysis of distance among elastically aligned EM maps, and results in visualizing those maps as points in a lower dimensional distance space.
-
subtract projection
- This protocol computes the subtraction between particles and a reference volume, by computing its projections with the same angles that input particles have. Then, each particle and the correspondent projection of the reference volume are numerically adjusted and subtracted using a mask which denotes the region to keep.
-
swarm consensus
- This is a 3D refinement protocol whose main input is a set of volumes and a set of particles. The set of particles has to be at full size (the finer sampling rate available), but the rest of inputs (reference volume and masks) can be at any downsampling factor. The protocol scales the input images and volumes to a size that depends on the target resolution. The input set of volumes is considered to be a swarm of volumes and they try to optimize the correlation between the volumes and the set of particles. This is an stochastic maximization and only a fraction of the particles are used to update the volumes and evaluate them.
-
tilt analysis
- Estimate the tilt of a micrograph, by analyzing the PSD correlations of different segments of the image.
-
tilt pairs particle picking (Particle picking)
- Picks particles in a set of untilted-tilted pairs of micrographs.
-
trigger data (Tools)
- Waits until certain number of images is prepared and then send them to output. It can be done in 3 ways: - If "Send all items to output?" is _No_: Once the number of items is reached, a setOfImages is returned and the protocol finishes (ending the streaming from this point). - If "Send all items to output?" is _Yes_ and: - If "Split items to multiple sets?" is _Yes_: Multiple closed outputs will be returned as soon as the number of items is reached. - If "Split items to multiple sets?" is _No_: Only one output is returned and it is growing up in batches of a certain number of items (completely in streaming).
-
validate fsc-q
- The protocol assesses the quality of the fit.
-
validate overfitting
- Check how the resolution changes with the number of projections used for 3D reconstruction. NOTE: Using the output plot, with the reconstruction of aligned gaussian noise, you can assess the validity of the reconstruction from your micrograph images. Practically, if the resolution of reconstruction based on your images is not considerably different from aligned gaussian noise one (for less number of particles),your images may not produce a valid reconstruction. This method has been proposed by: B. Heymann "Validation of 3D EM Reconstructions", 2015. (see References)
-
validate_nontilt
- Ranks a set of volumes according to their alignment reliability obtained from a clusterability test.
-
volume consensus
- This protocol performs a fusion of all the input volumes, which should be preprocessed with protocol 'volume substraction' saving volume 2, in order to be as similar as possible before the fusion. The output of this protocol is the consensus volume and another volume which indicates the maximun difference between input volumes in each voxel.
-
volume deform - Zernike3D
- Protocol for volume deformation based on Zernike3D.
-
volumes adjust
- This protocol scales a volume in order to assimilate it to another one. The volume with the best resolution should be the first one. The volumes should be aligned previously and they have to be equal in size
-
volumes subtraction
- This protocol scales a volume in order to adjust it to another one. Then, it can calculate the subtraction of the two volumes. Second input can be a pdb. The volumes should be aligned previously and they have to be equal in size
Contributors:






 Xmipptomo
Xmipptomo
Available methods:
-
Astigmatism rotation
- Rotate the astigmatism of a set of ctf tilt-series estimation given a set of transformation matrices comming from a set of tilt series.
-
Filter coordinates by map
- Filter coordinate by map both given a mask or a resolucion map from a tomogram
-
align transformations
- Protocol to rotate a series of alignments to a common reference defined by a Subtomogram Average
-
apply alignment subtomo
- Apply alignment matrix and produce a new setOfSubtomograms, with each subtomogram aligned to its reference.
-
apply alignment tilt-series
- Compute the interpolated tilt-series from its transform matrix.
-
cltomo
- Averages a set of subtomograms taking into account the missing edge.
-
connected components
- This protocol takes a set of coordinates and identifies connected components among the picked particles.
-
connected components to ROIs
- This protocol adjust a SetOfCoordinates (which usually will come from a connected componnent) to a ROI (region of interest) previously defined
-
crop tomograms
- Protocol to crop tomograms using xmipp_transform_window. The protocol allows to change the size of a tomogram/s, by removing the borders defined by the users
-
fit vesicles
- This protocol adjust a SetOfSubtomograms with coordinates assigned or a SetOfCoordinates3D, to a vesicle (ellipsoid), defining regions of interest (SetOfMeshes) for each vesicle as output.
-
half maps
- Create half maps from a SetOfSubtomograms and its alignment
-
imagej roi
- Tomogram ROI selection in IJ
-
local Resolution MonoTomo
- Given a tomogram the protocol assigns local resolutions to each voxel of the tomogram. To do that, thje protocol makes use of two half tomograms, called odd and even. These tomograms are reconstructed with the same alignment parameter but using the half of the data. For instance, the odd/even-images of the tilt series, or much better usign the odd/even frames of the movies (recommended). The result is a tomogram with the values of local resolution.
-
map back subtomos
- This protocol takes a tomogram, a reference subtomogram and a metadata with geometrical parameters (x,y,z) and places the reference subtomogram on the tomogram at the designated locations (map back). It has different representation options.
-
misalign tilt-series
- Introduce misalignment in the transformation matrix of a tilt-series. NOTE: The Interpolated tilt series in this case resembles a not aligned tilt series or an aligned one in case you want to apply the inverse of the misalignment transformation matrix.
-
phantom create subtomo
- Create subtomogram phantoms
-
phantom tomograms
- Create phantom tomograms with phantom particles and its coordinates with the right Scipion transformation matrix
-
resize tilt-series
- Wrapper protocol to Xmipp image resize applied on tilt-series
-
resize tomograms
- Protocol to to resize tomograms using xmipp_image_resize. The protocol allows to change the size of a tomogram/s by means of different methods
-
score transformations
- Protocol to score a series of alignments stored in a SetOfSubtomograms by quaternion distance analysis
-
score/filter coordinates
- Scoring and (optional) filtering of coordinates based on different scoring functions (carbon distance, neighbour distance)
-
split tilt-series
- Wrapper protocol to Xmipp split Odd Even on tilt-series
-
subtomo projection
- Project a set of volumes or subtomograms to obtain their X, Y or Z projection of the desired range of slices.
-
subtomo subtraction
- This protocol subtracts a subtomogram average to a SetOfSubtomograms, which are internally aligned and numerically adjusted in order to obtain reliable results. The adjustment and subtraction is perfomed by xmipp_volume_subtraction program. A mask can be provided if the user wants to perform the subtraction in a determined region.
-
tiltseries FlexAlign
- Simple protocol to average TiltSeries movies as basic motion correction. It is used mainly for testing purposes.
Contributors:
cryoCARE
Available methods:
-
CryoCARE Load Model
- Use two data-independent reconstructed tomograms to train a 3D cryo-CARE network.
-
CryoCARE Load Training Data
- Use two data-independent reconstructed tomograms to train a 3D cryo-CARE network.
-
CryoCARE Prediction
- Generate the final restored tomogram by applying the cryoCARE trained network to bothtomograms followed by per-pixel averaging.
-
CryoCARE Training
- Use two data-independent reconstructed tomograms to train a 3D cryo-CARE network.
-
CryoCARE Training Data Extraction
- Operate the data to make it be expressed as expected by cryoCARE net.
Contributors:

emantomo
Available methods:
-
align tilt series
- This protocol wraps *e2tomogram.py* EMAN2 program. Alignment of the tilt-series is performed iteratively.
-
average subtomo
- This protocol wraps *e2spt_average.py* EMAN2 program. It computed the average from a SetOfSubtomograms based on their alignment.
-
classify subtomos
- This protocol wraps *e2spt_classify.py* EMAN2 program. Protocol to performs a conventional iterative subtomogram averaging using the full set of particles. It will take a set of subtomograms (particles) and a set of Averages(reference) and 3D reconstruct a subtomogram. It also builds a set of subtomograms that contains the original particles plus the score, coverage and align matrix per subtomogram .
-
ctf estimation (CTF estimation)
- Protocol for CTF estimation from tilt series using e2spt_tomoctf.py
-
extraction from tilt-series
- Extraction of subtomograms from tilt serie using EMAN2 e2spt_extract.py.
-
extraction from tomogram
- Extraction for Tomo. Uses EMAN2 e2spt_boxer_old.py.
-
resize tomos
- Tomogram resize using e2proc3d
-
sub-tilt refinement
- Sub-tilt refinement from Eman e2spt_tiltrefine.py
-
subtomogram refinement (3D refinement)
- This protocol wraps *e2spt_refine.py* EMAN2 program. Protocol to performs a conventional iterative subtomogram averaging using the full set of particles. It will take a set of subtomograms (particles) and a subtomogram(reference, potentially coming from the initial model protocol) and 3D reconstruct a subtomogram. It also builds a set of subtomograms that contains the original particles plus the score, coverage and align matrix per subtomogram .
-
template matching
- This protocol wraps *e2spt_tempmatch.py* EMAN2 program. It will perform a sweep of an initial volume against a tomogram to find correlation peaks and extract the corresponding subtomogram coordinates
-
tomo boxer (Particle picking)
- Manual picker for Tomo. Uses EMAN2 e2spt_boxer.py.
-
tomo boxer convnet
- Eman Deep Learning based picking for Tomography
-
tomo fill mw
- This protocol is a wrapper of *e2tomo_mwfill*. This program can be used to fill in the missing wedge of a SetOfTomograms with useful information based on a neural network
-
tomo initial model (Initial model)
- This protocol wraps *e2spt_sgd.py* EMAN2 program. It will take a set of subtomograms (particles) and a subtomogram(reference) and build a subtomogram suitable for use as initial models in tomography. It also builds a set of subtomograms that contains the original particles plus the score, coverage and align matrix per subtomogram .
-
tomo reconstruction (3D reconstruction)
- This protocol wraps *e2tomogram.py* EMAN2 program. Tomogram reconstruction from aligned tilt series. Tomograms are not normally reconstructed at full resolution, generally limited to 1k x 1k or 2k x 2k, but the tilt-series are aligned at full resolution. For high resolution subtomogram averaging, the raw tilt-series data is used, based on coordinates from particle picking in the downsampled tomograms. On a typical workstation reconstruction takes about 4-5 minutes per tomogram.
Contributors:
gCTF
Available methods:
-
ctf estimation (CTF estimation)
- Estimates CTF on a set of micrographs using Gctf. To find more information about Gctf go to: https://www2.mrc-lmb.cam.ac.uk/research/locally-developed-software/zhang-software/#gctf
-
ctf refinement (CTF estimation)
- Refines local CTF of a set of particles using Gctf. To find more information about Gctf go to: https://www2.mrc-lmb.cam.ac.uk/research/locally-developed-software/zhang-software/#gctf
-
tilt-series gctf (CTF estimation)
- CTF estimation on a set of tilt series using GCTF.
Contributors:
 prody2
prody2
This plugin provide a wrapper around ProDy software: A Python Package for Protein Dynamics Analysis
Plugin url https://github.com/scipion-em/scipion-em-prody.
Available in Linux. Scientific software, Image processing, cryo emAvailable methods:
-
ANM analysis
- This protocol will perform normal mode analysis (NMA) using the anisotropic network model (ANM)
-
Atom Alignment
- This protocol will perform atomic structure mapping and superposition
-
Atom Selection
- This protocol will perform atom selection
-
Compare modes
- This protocol will compare two SetOfNormalModes objects
-
Deformation
- This protocol will perform deformation vector analysis
-
Domain Decomposition
- This protocol will perform dynamical domain decomposition
-
Edit modes
- This protocol will edit a SetOfNormalModes object to have more or fewer nodes
-
GNM analysis
- This protocol will perform normal mode analysis (NMA) using the Gaussian network model (GNM)
-
Import ensemble
- This protocol will import a ProDy Ensemble or PDBEnsemble
-
Import modes
- This protocol will import ProDy modes to a SetOfNormalModes object
-
PCA
- This protocol will perform ProDy principal component analysis (PCA) using atomic structures
-
Projection
- This module will provide ProDy projection of structural ensembles on principal component or normal modes
-
RTB analysis
- This protocol will perform normal mode analysis (NMA) using the rotation and translation of blocks (RTB) framework
-
buildPDBEnsemble
- This protocol will build a PDBEnsemble
Contributors:
pwed
scipion-ed is the base plugin defining the Domain for Electron Diffraction image processing
Plugin url https://github.com/scipion-ed/scipion-ed.
Available in Linux. Scientific software, Image processing, cryo emAvailable methods:
-
Base class for other ed Import protocols. (Import)
- Base class for other ed Import protocols.
Contributors:
pwem
Base plugin to deal with EM data. Provides all the import protocols and the model for EM objects (Micrograpths, Movies, Volumes, etc)
Plugin url https://github.com/scipion-em/scipion-em.
Available in Linux. Scientific software, Image processing, cryo emAvailable methods:
-
Manual subset (Tools)
- Create subsets from the GUI. This protocol will be executed mainly calling the script 'pw_create_image_subsets.py' from the ShowJ gui. The enabled/disabled changes will be stored in a temporary sqlite file that will be read to create the new subset.
-
ProtCreateFSC
- Description missing, might be a new development.
-
ProtCreateMask (Masking)
- Description missing, might be a new development.
-
ProtParticles
- test
-
assign Orig & Sampling
- Modify the origin and sampling values assigned to a 3D map
-
assign alignment
- Assign a the alignment calculated for a set of particles to another set. This protocol will take into account the differences of pixel size (A/pix) between the two sets and multiply by the right factor the shifts. The particles with the alignment can also be a subset of the other images
-
assign ctf
- This protocol assigns a CTF estimation to a particular set of particles producing a new set.
-
assign gain to movies
- Assign a gain image to a set of movies
-
average frames
- Very simple protocol to align all the frames of a given data collection session. It can be used as a sanity check.
-
box size related parameters
- Protocol to make mathematical operations on particle picking boxsize. For sanity check all the generated outputs are even numbers.
-
classes consensus
- Compare two SetOfClasses
-
create stream data
- create setofXXXX in streaming mode. micrograph -> read a micrograph in memory and writes it nDim times movie -> read a movie in memory and writes it nDim times randomMicrographs -> creates a micrograph with random values and applies a random CTF particles -> read nDim particles in memory and writes it in streaming
-
edit set
- Protocol to edit attributes of all the items of a set using a formula. This could be useful for editing some values in the set. Use this protocol with extreme care, you can easily produce a set that is not consistent. Some predefined operation are offered as given a set of particles with projection alignment modify the alignment matrix so when reconstructed the volume is rotated.
-
export emdb (Export)
- generates files for volumes and FSCs to submit structures to EMDB
-
export to emdb/pdb (Export)
- generates files for elements to submit structures to EMDB/PDB. Since mmcif/pdb is only partially supported by some software the protocol creates 4 versions of the atomic struct file with the hope that at least one of them will work.
-
extract coordinates
- Extract the coordinates information from a set of particles. This protocol is useful when we want to re-extract the particles (maybe resulting from classification and cleaning) with the original dimensions. It can be also handy to visualize the resulting particles in their location on micrographs.
-
filter set
- Protocol to filter sets based on its attributes through an expression that should evaluate to true or false. Some predefined expresions are stored (i.e. distance to center, distance between coordinates)
-
import atomic structure (Import)
- Protocol to import an atomic structure to the project.Format may be PDB or MMCIF
-
import averages (Import)
- Protocol to import a set of averages to the project
-
import coordinate pairs (Import)
- Protocol to import a set of tilt pair coordinates
-
import coordinates (Import)
- Protocol to import a set of coordinates
-
import ctf (Import)
- Common protocol to import a set of ctfs into the project
-
import mask (Import)
- Class for import masks from existing files.
-
import micrographs (Import)
- Protocol to import a set of micrographs to the project
-
import movies (Import)
- Protocol to import a set of movies (from direct detector cameras) to the project.
-
import particles (Import)
- Protocol to import a set of particles to the project
-
import sequence (Import)
- Protocol to import an aminoacid/nucleotide sequence file to the project
-
import set of atomic structures (Import)
- Protocol to import a set of atomic structure to the project. Format may be PDB or MMCIF
-
import tilted micrographs (Import)
- Protocol to import untilted-tilted pairs of micrographs in the project
-
import volumes (Import)
- Protocol to import a set of volumes to the project
-
invert hand
- Modify the transformation matrix of a set of particles So that the handedness changes
-
join sets (Tools)
- Protocol to join two or more sets of images. This protocol allows to select two or more set of images and will produce another set joining all elements of the selected sets. It will validate that all sets are of the same type of elements (Micrographs, Particles or Volumes)
-
manual check point
- This protocol is kept running for a time determined by a parameter or until the user determines it is convenient. This protocol is useful if we want a given protocol to be launched at the time the user sees appropriate.
-
mathematical operator
- Protocol to make mathematical operations on different inputs
-
metadata editor
- Protocol to edit attributes of all the items of a set using a formula. This could be useful for corrupting your data for testing purposes or editing some values in the set that make sense to do it. Use this with extreme care, you can easily ruin your processing.
-
movie eraser
- It will REMOVE movies that already have been aligned into micrographs. WARNING: There is no way back. Be sure you understand the consequences.
-
numeric classes extractor
- Extracts items from a SetOfClasses based on number of items assigned to the class
-
parallel test
- A parallel test protocol.
-
particles subset by coordinates
- Create a subset of those particles that have a particular set of coordinates
-
particles subset by micrograph (Tools)
- Create a subset of those particles that come from a particular set of micrographs
-
pdf report
- Produce a pdf report from the files in a given directory. Supported file formats: *.tex, *.txt, *.jpg, *.png, *.pdf Files in the directory are sorted by name alphabetically, so if you want them to have the right order a possibility is to name them as 0010-myText.txt 0020-aFigure.png 0030-anotherFigure.jpg 0040-aPaper.pdf 0050-anotherText.tex ... when these files are sorted, they will be sorted by the number in front.
-
picking difference
- Protocol to compute the difference between a reference SetOfParticles and a another set (usually a negative reference). The output will be a SetOfCoordinates with the particles in the reference input that are not close to coordinates in the negative input.
-
split sets (Tools)
- Protocol to split a set in two or more subsets.
-
stress
- stress will stress test a computer system in various selectable ways. Several options require the program stress-ng.
-
subset (Tools)
- Create a set with the elements of an original set that are also referenced in another set. Usually there is a bigger set with all the elements, and a smaller one obtained from classification, cleaning, etc. The desired result is a set with the elements from the original set that are also present somehow in the smaller set (in the smaller set they may be downsampled or processed in some other way). Both sets should be of the same kind (micrographs, particles, volumes) or related (micrographs and CTFs for example).
Contributors:
pyworkflowtests
Available methods:
-
test output
- Protocol to test scalar output and input linking
Contributors:
tomo3D
Available methods:
-
denoise tomogram
- Denoises sets of tomograms using methods described in https://sites.google.com/site/3demimageprocessing/ Returns the set of denoised tomograms
-
reconstruct tomogram
- Reconstruct tomograms from aligned tilt series using TOMO3D from Software from: https://sites.google.com/site/3demimageprocessing/ Returns the set of tomograms
Contributors:
tomoviz
Available methods:
-
filter by normal
- This protocol takes surfaces or ROIs (SetOfMeshes) and a SetOfSubtomograms or SetOfCoordinates3D with transformation matrix and filters them by different criteria related with the normal direction.
-
picking consensus
- Protocol to estimate the agreement between different particle picking algorithms. The protocol takes several Sets of Coordinates calculated by different programs and/or different parameter settings. Let's say: we consider N independent pickings. Then, a coordinate is considered to be a correct particle if M pickers have selected the same particle (within a radius in pixels specified in the form). If you want to be very strict, then set M=N; that is, a coordinate represents a particle if it has been selected by all particles (this is the default behaviour). Then you may relax this condition by setting M=N-1, N-2, ... If you want to be very flexible, set M=1, in this way it suffices that 1 picker has selected the coordinate to be considered as a particle. Note that in this way, the cleaning of the dataset has to be performed by other means (screen particles, 2D and 3D classification, ...).
-
remove duplicates
- This protocol removes coordinates that are closer than a given threshold. The remaining coordinate is the average of the previous ones.